Raksta medicīnas eksperts

Jaunas publikācijas
Cistiskā fibroze
Pēdējā pārskatīšana: 04.07.2025

Visi iLive saturs ir medicīniski pārskatīts vai pārbaudīts, lai nodrošinātu pēc iespējas lielāku faktisko precizitāti.
Mums ir stingras iegādes vadlīnijas un tikai saikne ar cienījamiem mediju portāliem, akadēmiskām pētniecības iestādēm un, ja vien iespējams, medicīniski salīdzinošiem pārskatiem. Ņemiet vērā, ka iekavās ([1], [2] uc) esošie numuri ir klikšķi uz šīm studijām.
Ja uzskatāt, ka kāds no mūsu saturiem ir neprecīzs, novecojis vai citādi apšaubāms, lūdzu, atlasiet to un nospiediet Ctrl + Enter.
Cistiskā fibroze ir ģenētiska autosomāli recesīva monogēna slimība, kurai raksturīgi dzīvībai svarīgu orgānu eksokrīno dziedzeru sekrēcijas traucējumi ar bojājumiem galvenokārt elpošanas un gremošanas sistēmās, smaga gaita un nelabvēlīga prognoze.
 [ 1 ]
[ 1 ]
Epidemioloģija
Cistiskās fibrozes sastopamība svārstās no 1:2500 līdz 1:4600 jaundzimušajiem. Katru gadu pasaulē piedzimst aptuveni 45 000 cilvēku ar cistisko fibrozi. Cistiskās fibrozes gēna nesēju sastopamība ir 3–4%, un aptuveni 275 miljoni cilvēku visā pasaulē ir šī gēna nesēji, no kuriem aptuveni 5 miljoni dzīvo Krievijā un aptuveni 12,5 miljoni NVS valstīs.
Cēloņi cistiskā fibroze
Cistiskā fibroze tiek pārnesta autosomāli recesīvā veidā. Cistiskās fibrozes gēns atrodas 7. autosomā, satur 27 eksonus un sastāv no 250 000 nukleotīdu pāriem.
Vienam gēnam var būt daudz mutāciju, no kurām katra ir raksturīga konkrētai populācijai vai ģeogrāfiskam reģionam. Ir aprakstītas vairāk nekā 520 mutācijas, no kurām visizplatītākā ir delta-P-508, t.i., aminoskābes fenilalanīna aizvietošana 508. pozīcijā.
Pathogenesis
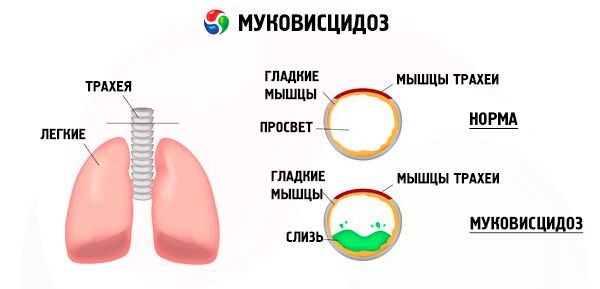
Mutācijas cistiskās fibrozes gēnā izjauc proteīna, ko sauc par CFTR (cistiskā fibroze transmembrāna regulators), struktūru un funkciju. Šis proteīns darbojas kā hlorīda kanāls, kas iesaistīts bronhopulmonālās sistēmas, kuņģa-zarnu trakta, aizkuņģa dziedzera, aknu un reproduktīvās sistēmas epitēlija šūnu ūdens un elektrolītu apmaiņā. CFTR proteīna funkcijas un struktūras traucējumu rezultātā šūnas iekšpusē uzkrājas hlorīda joni Cl⁻ . Tas noved pie elektriskā potenciāla izmaiņām izvadkanālu lūmenā, kas atvieglo liela daudzuma nātrija jonu (Na⁺) plūsmu no kanāla lūmena šūnā un vēl vairāk uzlabo ūdens uzsūkšanos no pericelulārās telpas.
Šo izmaiņu rezultātā vairuma eksokrīno dziedzeru sekrēcija sabiezē, tiek traucēta tās evakuācija, kas izraisa izteiktus sekundārus traucējumus orgānos un sistēmās, visizteiktāko bronhopulmonālajā un gremošanas sistēmā.
Bronhos attīstās hronisks dažādas intensitātes iekaisuma process, strauji tiek traucēta ciliārā epitēlija funkcija, krēpas kļūst ļoti viskozas, biezas, ļoti grūti evakuējamas, novērojama to stagnācija, veidojas bronhiolo- un bronhektāzes, kas laika gaitā kļūst arvien biežākas. Šīs izmaiņas izraisa hipoksijas palielināšanos un hroniskas plaušu sirds slimības veidošanos.
Pacientiem ar cistisko fibrozi ir ārkārtīgi liela predispozīcija hroniska iekaisuma attīstībai bronhopulmonālajā sistēmā. Tas ir saistīts ar izteiktiem vietējās bronhopulmonālās aizsardzības sistēmas traucējumiem (samazināts IgA, interferona līmenis, alveolāro makrofāgu un leikocītu fagocitārā funkcija).
Alveolārajiem makrofāgiem ir liela nozīme hroniska iekaisuma attīstībā bronhopulmonālajā sistēmā. Tie ražo lielu daudzumu IL-8, kas dramatiski palielina neitrofilu hemotaksi bronhu kokā. Neitrofili lielos daudzumos uzkrājas bronhos un kopā ar epitēlija šūnām izdala daudzus iekaisumu veicinošus citokīnus, tostarp IL-1, 8, 6, audzēja nekrozes faktoru un leikotriēnus.
Svarīga loma bronhopulmonālās sistēmas bojājumu patoģenēzē ir arī fermenta elastāzes augstajai aktivitātei. Izšķir eksogēnu un endogēnu elastāzi. Pirmo ražo baktēriju flora (īpaši Pseudomonas aeruginosa), otro - neitrofilie leikocīti. Elastāze iznīcina epitēliju un citus bronhu strukturālos elementus, kas veicina turpmāku mukociliārā transporta traucējumus un strauju bronhektāzes veidošanos.
Neitrofilie leikocīti izdala arī citus proteolītiskos enzīmus. Alfa-1-antipirsīns un leikoproteāžu sekrēcijas inhibitors neitralizē proteolītisko enzīmu ietekmi un tādējādi aizsargā bronhopulmonālo sistēmu no to kaitīgās ietekmes. Tomēr diemžēl pacientiem ar cistisko fibrozi šie aizsargfaktori tiek nomākti ar ievērojamu neitrofilo proteāžu daudzumu.
Visi šie apstākļi veicina infekcijas iekļūšanu bronhopulmonālajā sistēmā un hroniska strutaina bronhīta attīstību. Turklāt jāņem vērā, ka cistiskās fibrozes gēna kodētais defektīvais proteīns maina bronhu epitēlija funkcionālo stāvokli, kas veicina baktēriju, galvenokārt Pseudomonas aeruginosa, adhēziju pie bronhu epitēlija.
Līdztekus bronhopulmonālās sistēmas patoloģijai, cistiskā fibroze izraisa arī smagus bojājumus aizkuņģa dziedzerim, kuņģim, lielajai un tievajai zarnai, aknām.
Simptomi cistiskā fibroze
Cistiskā fibroze izpaužas ar dažādiem klīniskiem simptomiem. Jaundzimušajiem slimība var izpausties ar mekonija ileusu. Tripsīna trūkuma vai pat pilnīgas neesamības dēļ mekonijs kļūst ļoti blīvs, viskozs un uzkrājas ileocekālajā rajonā. Tālāk attīstās zarnu nosprostojums, kas izpaužas kā intensīva vemšana ar žults piejaukumu, vēdera uzpūšanās, mekonija izdalīšanās trūkums, peritonīta simptomu attīstība un strauja smagas intoksikācijas sindroma klīnisko izpausmju pastiprināšanās. Bērns var nomirt pirmajās dzīves dienās, ja netiek veikta steidzama ķirurģiska iejaukšanās.
Mazāk smagos gadījumos raksturīga cistiskās fibrozes pazīme ir bagātīga, bieža vēdera izeja, taukaina, ar lielu tauku daudzumu, ar ļoti nepatīkamu smaku. 1/3 pacientu tiek novērota taisnās zarnas noslīdēšana.
Pēc tam pacientiem turpina rasties zarnu darbības traucējumi, malabsorbcijas sindroms, smagi fiziskās attīstības traucējumi un smaga hipovitaminoze.
Pirmajā vai otrajā dzīves gadā parādās bronhopulmonālās sistēmas bojājuma simptomi (viegla slimības forma), kas izpaužas kā klepus, kas var būt ārkārtīgi izteikts un atgādināt klepu ar garo klepu. Klepu pavada cianoze, elpas trūkums un biezu krēpu atdalīšanās, sākotnēji gļotainu, bet pēc tam strutainu. Pakāpeniski veidojas hroniska obstruktīva bronhīta un bronhektāzes, plaušu emfizēmas un elpošanas mazspējas klīniskā aina. Bērni ir ārkārtīgi uzņēmīgi pret akūtām elpceļu vīrusu un baktēriju infekcijām, kas veicina bronhopulmonālās patoloģijas saasināšanos un progresēšanu. Iespējama infekcijas atkarīgas bronhiālās astmas attīstība.
Skolas vecuma bērniem cistiskā fibroze var izpausties kā "zarnu kolikas". Pacienti sūdzas par stiprām paroksizmālām sāpēm vēderā, vēdera uzpūšanos un atkārtotu vemšanu. Palpējot vēderu, tiek noteikti blīvi veidojumi, kas atrodas resnās zarnas projekcijā - fekāliju masas, kas sajauktas ar biezām, blīvām gļotām. Bērni ir ļoti pakļauti hipohlorēmiskās alkalozes attīstībai pārmērīgas sāls izdalīšanās dēļ ar sviedriem karstā laikā, savukārt uz bērna ādas parādās "sāls sals".
Bronhopulmonālās sistēmas traucējumi pieaugušajiem
Bronhopulmonālās sistēmas bojājumiem pacientiem ar cistisko fibrozi (slimības plaušu forma) raksturīga hroniska strutaina obstruktīva bronhīta, bronhektāzes, hroniskas pneimonijas, plaušu emfizēmas, elpošanas mazspējas un plaušu sirds slimības attīstība. Dažiem pacientiem attīstās pneimotorakss un citas cistiskās fibrozes komplikācijas: atelektāze, plaušu abscesi, hemoptīze, plaušu asiņošana un infekcijas atkarīga bronhiālā astma.
Pacienti sūdzas par sāpīgu paroksizmālu klepu ar ļoti viskozu, grūti atdalāmu gļotainu krēpu, dažreiz ar asiņu piejaukumu. Turklāt ārkārtīgi raksturīga ir elpas trūkums, vispirms fiziskas slodzes laikā, bet pēc tam miera stāvoklī. Elpas trūkumu izraisa bronhu obstrukcija. Daudzi pacienti sūdzas par hronisku rinītu, ko izraisa polipoze un sinusīts. Raksturīgs ir arī ievērojams vājums, progresējoša darbspēju samazināšanās, biežas akūtas elpceļu vīrusu slimības. Izmeklējot, uzmanība tiek pievērsta bālai ādai, sejas pietūkumam, redzamo gļotādu cianozei un smagai elpas trūkumam. Attīstoties dekompensētai plaušu sirds slimībai, kājās parādās tūska. Var novērot pirkstu gala falangu sabiezēšanu bungu stilbiņu veidā un nagus pulksteņstiklu veidā. Krūškurvis iegūst mucveida formu (plaušu emfizēmas attīstības dēļ).
Plaušu perkusija atklāj emfizēmas pazīmes – kastveida skaņu, asu plaušu malas kustīguma ierobežojumu un plaušu apakšējās robežas pazemināšanos. Plaušu auskultācija atklāj raupju elpošanu ar pagarinātu izelpu, izkliedētu sausu sēkšanu un mitru, vidēju un smalku burbuļojošu sēkšanu. Smagas plaušu emfizēmas gadījumā elpošana ir strauji pavājināta.
Cistiskās fibrozes ekstrapulmonālās izpausmes
Cistiskās fibrozes ekstrapulmonālās izpausmes var būt diezgan izteiktas un rodas bieži.
Aizkuņģa dziedzera bojājumi
85% pacientu ar cistisko fibrozi novēro dažādas smaguma pakāpes aizkuņģa dziedzera eksokrīnās funkcijas nepietiekamību. Ar nelieliem aizkuņģa dziedzera bojājumiem maldigestija un malabsorbcijas sindromi nav, ir tikai laboratoriskas eksokrīnās nepietiekamības izpausmes (zems tripsīna un lipāzes līmenis asinīs un divpadsmitpirkstu zarnas saturā; bieži smaga steatoreja). Ir zināms, ka, lai novērstu maldigestija sindromu, pietiek ar tikai 1 līdz 2% kopējās lipāzes sekrēciju. Klīniski izpaužas tikai būtiski eksokrīnās funkcijas traucējumi.
Normālos apstākļos aizkuņģa dziedzera acini izdala šķidru sekrētu, kas bagāts ar enzīmiem. Sekrētam pārvietojoties pa izvadkanālu, tas bagātinās ar ūdeni un anjoniem, un tas kļūst vēl šķidrāks. Cistiskās fibrozes gadījumā transmembrānas regulatora (hlorīda kanāla) struktūras un funkcijas traucējumu dēļ aizkuņģa dziedzera sekrēts nesaņem pietiekamu daudzumu šķidruma, tas kļūst viskozs, un tā kustības ātrums pa izvadkanālu strauji palēninās. Sekrēta olbaltumvielas nogulsnējas uz mazo izvadkanālu sieniņām, kā rezultātā tie tiek aizsprostoti. Slimībai progresējot, galu galā attīstās acini destrukcijas un atrofijas process - veidojas hronisks pankreatīts ar eksokrīnu aizkuņģa dziedzera mazspēju. Tas klīniski atspoguļojas malgremošanas un malabsorbcijas sindromu attīstībā. Aizkuņģa dziedzera mazspēja ir galvenais tauku malabsorbcijas cēlonis cistiskās fibrozes gadījumā, taču to parasti novēro ar ievērojamu lipāzes deficītu. Foršers un Durijs (1991) norāda, ka pilnīgas aizkuņģa dziedzera lipāzes neesamības gadījumā tauki tiek sadalīti un absorbēti par 50–60 %, kas ir saistīts ar kuņģa un siekalu (sublingvālo) lipāžu klātbūtni, kuru aktivitāte ir tuvu normas apakšējai robežai. Līdz ar tauku sadalīšanās un absorbcijas traucējumiem tiek traucēta arī olbaltumvielu sadalīšanās un reabsorbcija. Apmēram 50 % no ar pārtiku uzņemtajām olbaltumvielām tiek zaudēti ar fekālijām. Ogļhidrātu absorbcija tiek ietekmēta mazākā mērā, neskatoties uz α-amilāzes deficītu, taču ogļhidrātu metabolisms var tikt ievērojami traucēts.
Aizkuņģa dziedzera bojājumi izpaužas kā maldigestija un malabsorbcijas sindroma attīstība ar ievērojamu svara zudumu un bagātīgu taukainu izkārnījumu daudzumu.
Arī gremošanas traucējumu un malabsorbcijas sindromu attīstību veicina smaga zarnu dziedzeru disfunkcija, zarnu sulas sekrēcijas traucējumi un zarnu enzīmu satura samazināšanās tajā.
Gremošanas traucējumu un malabsorbcijas sindromus sauc arī par cistiskās fibrozes zarnu formu.
Pacientiem ar cistisko fibrozi slimības vēlīnā stadijā (2% bērnu un 15% pieaugušo) novēro aizkuņģa dziedzera endokrīnās sistēmas darbības traucējumus (cukura diabētu).
[ 16 ], [ 17 ], [ 18 ], [ 19 ]
Aknu un žultsceļu bojājumi
13% pacientu ar jauktu un zarnu cistiskās fibrozes formu attīstās aknu ciroze. Vistipiskāk tā ir saistīta ar mutācijām W128X, delta-P508 un X1303K. Aknu biliārā ciroze ar portālu hipertensiju tiek konstatēta 5-10% pacientu. Saskaņā ar Welch, Smith (1995) datiem, klīniskās, morfoloģiskās, laboratoriskās, instrumentālās aknu bojājumu pazīmes tiek konstatētas 86% pacientu ar cistisko fibrozi.
Daudziem pacientiem ar cistisko fibrozi attīstās arī hronisks holecistīts, bieži vien ar kalculozi.
Dzimumdziedzeru disfunkcija
Pacientiem ar cistisko fibrozi var rasties azoospermija, kas ir neauglības cēlonis. Samazināta auglība ir raksturīga arī sievietēm.
Posmi
Plaušu cistiskās fibrozes gadījumā ir trīs smaguma pakāpes.
Vieglu cistiskās fibrozes formu raksturo reti paasinājumi (ne vairāk kā reizi gadā); remisijas periodos klīniskās izpausmes praktiski nav, un pacienti spēj strādāt.
Vidēja smaguma pakāpe - paasinājumi tiek novēroti 2-3 reizes gadā un ilgst apmēram 2 mēnešus vai ilgāk. Paasinājuma fāzē ir intensīvs klepus ar grūti atdalāmu krēpu, elpas trūkums pat pie nelielas fiziskas slodzes, subfebrīla ķermeņa temperatūra, vispārējs vājums, svīšana. Vienlaikus ir aizkuņģa dziedzera eksokrīnās funkcijas pārkāpums. Remisijas fāzē darbspējas netiek pilnībā atjaunotas, elpas trūkums fiziskas slodzes laikā saglabājas.
Smagu gaitu raksturo ļoti biežas slimības paasināšanās. Remisijas praktiski nav. Klīniskajā ainā priekšplānā izvirzās smaga elpošanas mazspēja, hroniskas plaušu sirds slimības simptomi, bieži dekompensēti, raksturīga hemoptīze. Novērots ievērojams svara zudums, pacienti kļūst pilnībā invalīdi. Parasti smagu bronhopulmonālu patoloģiju pavada izteikta aizkuņģa dziedzera disfunkcija.
Veidlapas
- Bronhopulmonāli bojājumi
- Atkārtota un atkārtota pneimonija ar ilgstošu gaitu.
- Abscesējoša pneimonija, īpaši zīdaiņiem.
- Hroniska pneimonija, īpaši divpusēja.
- Bronhiālā astma, kas ir rezistenta pret tradicionālo terapiju.
- Recidivējošs bronhīts, bronhiolīts, īpaši ar Pseudomonas aeruginosa kultūru.
- Izmaiņas kuņģa-zarnu traktā
- Mekonija ileuss un tā ekvivalenti.
- Nezināmas ģenēzes zarnu absorbcijas traucējumu sindroms.
- Obstruktīva dzelte jaundzimušajiem ar ilgstošu gaitu.
- Aknu ciroze.
- Cukura diabēts.
- Gastroezofageālais reflukss.
- Žultsakmeņu slimība.
- Taisnās zarnas prolapss.
- Izmaiņas citos orgānos un sistēmās
- Augšanas un attīstības traucējumi.
- Aizkavēta seksuālā attīstība.
- Vīriešu neauglība.
- Deguna polipi.
- Brāļi un māsas no ģimenēm ar cistisko fibrozi.
[ 24 ]
Komplikācijas un sekas
Komplikācijas no kuņģa-zarnu trakta ietver:
- Cukura diabēts attīstās 8–12% pacientu, kas vecāki par 25 gadiem.
- Fibrozējošā kolonopātija.
- Mekonija ileuss jaundzimušo periodā (12% jaundzimušo ar cistisko fibrozi), distālā zarnu obstrukcijas sindroms, taisnās zarnas prolapss, peptiska čūla un gastroezofageālā refluksa slimība.
Aknu komplikācijas:
- Taukainā aknu slimība (30–60 % pacientu),
- Fokālā biliārā ciroze, multinodulārā biliārā ciroze un ar tām saistītā portālā hipertensija.
Portāla hipertensija dažreiz izraisa nāvi barības vada varikozu vēnu dēļ.
Holecistīta un žultsakmeņu izplatība pacientiem ar cistisko fibrozi ir lielāka nekā citiem indivīdiem.
Aizkavēta pubertāte, samazināta auglība un citas komplikācijas. Lielākajai daļai vīriešu ir azoospermija un sēklas vads (sēklvadu) nepietiekama attīstība.
Diagnostika cistiskā fibroze
Vispārējā asins analīze - raksturīga dažādas smaguma pakāpes anēmija, parasti normo- vai hipohroma. Anēmijai ir polifaktoriāla ģenēze (samazināta dzelzs un B12 vitamīna uzsūkšanās zarnās malabsorbcijas sindroma attīstības dēļ). Iespējama leikopēnija, attīstoties strutainam bronhītam un pneimonijai - leikocitoze, paaugstināts ESR.
Vispārēja urīna analīze - nav būtisku izmaiņu, retos gadījumos novēro nelielu proteinūriju.
Koproloģiskā izmeklēšanā novēro steatoreju, kreatoreju. Bekers (1987) iesaka noteikt himotripsīna un taukskābju daudzumu fekālijās. Pirms himotripsīna noteikšanas fekālijās, vismaz 3 dienas pirms izmeklējuma jāpārtrauc gremošanas enzīmu lietošana. Cistiskās fibrozes gadījumā himotripsīna daudzums fekālijās ir samazināts, bet taukskābju daudzums ir palielināts (normāla taukskābju izdalīšanās ir mazāka par 20 mmol/dienā). Jāņem vērā, ka palielināta taukskābju izdalīšanās ar fekālijām tiek novērota arī:
- konjugēto taukskābju deficīts tievajās zarnās aknu mazspējas, žultsvadu obstrukcijas, ievērojamas tievās zarnas baktēriju kolonizācijas dēļ (šajā gadījumā notiek intensīva žultsskābju hidrolīze);
- ileīts;
- celiakija (ar malabsorbcijas sindroma attīstību);
- enterīts;
- zarnu limfomas;
- Vipla slimība;
- pārtikas alerģijas;
- Paātrināta pārtikas masu tranzīta gadījumā dažādas izcelsmes caurejas, karcinoīda sindroma, tireotoksikozes gadījumā.
Bioķīmiskā asins analīze - samazināts kopējais olbaltumvielu un albumīna līmenis, paaugstināts alfa2 un gamma globulīnu, bilirubīna un aminotransferāžu līmenis (aknu bojājuma gadījumā), samazināta amilāzes, lipāzes, tripsīna un dzelzs un kalcija līmeņa aktivitāte (maldigestio sindroma, malabsorbcijas attīstības gadījumā).
Krēpu analīze - liela skaita neitrofilo leikocītu un mikroorganismu klātbūtne (krēpu bakterioskopijas laikā).
Pētot tievās zarnas absorbcijas funkciju un aizkuņģa dziedzera eksokrīno funkciju, tiek atklāti ievērojami traucējumi.
Plaušu rentgena izmeklēšana - atklāj izmaiņas, kuru smagums ir atkarīgs no slimības smaguma pakāpes un fāzes. Raksturīgākās izmaiņas ir:
- palielināta plaušu struktūra peribronhiālu intersticiālu izmaiņu dēļ;
- plaušu sakņu paplašināšanās;
- plaušu lobulāras, subsegmentālas vai pat segmentālas atelektāzes attēls;
- palielināta plaušu lauku caurspīdība, galvenokārt augšējos sekcijās, diafragmas zemā pozīcija un nepietiekama kustīgums, retrosternālās telpas paplašināšanās (plaušu emfizēmas izpausme);
- segmentāla vai polisegmentāla plaušu audu infiltrācija (pneimonijas attīstībā).
Bronhogrāfijā atklājas izmaiņas, ko izraisa bronhu aizsprostojums ar viskozu krēpu (bronhu pildījuma fragmentācija ar kontrastu, nelīdzenas kontūras, bronhu plīsuma parādība, ievērojama sānu zaru skaita samazināšanās), kā arī bronhoekāzes (cilindriskas, jauktas), kas lokalizējas galvenokārt plaušu apakšējās daļās.
Bronhoskopija atklāj difūzu strutainu bronhītu ar bagātīgu biezu, viskozu krēpu un fibrīna plēvēm.
Spirometrija - jau slimības sākumposmā atklāj obstruktīva tipa elpošanas mazspēju (samazināts FVC, FEV1, Tiffno indekss), ierobežojošu (samazināts FVC) vai, visbiežāk, obstruktīvi ierobežojošu (samazināts FVC, FVC, FEV1, Tiffno indekss).
Gibsona un Kuka sviedru tests (sviedru elektrolītu tests) ietver svīšanas stimulēšanu, izmantojot pilokarpīna elektroforēzi, ar sekojošu hlorīdu noteikšanu sviedros. Doerehuks (1987) apraksta testu šādi. Pilokarpīna elektroforēzi veic uz apakšdelma, elektriskā strāva ir 3 mA. Pēc ādas attīrīšanas ar destilētu ūdeni sviedrus savāc, izmantojot filtrpapīru, kas uzlikts uz stimulētās zonas, pārklāts ar marli, lai novērstu iztvaikošanu. Pēc 30–60 minūtēm filtrpapīru noņem un eluē destilētā ūdenī. Tiek mērīts savākto sviedru daudzums. Lai iegūtu ticamus rezultātus, nepieciešams savākt vismaz 50 mg (vēlams 100 mg) sviedru.
Ja hlorīdu koncentrācija pārsniedz 60 mmol/l, cistiskās fibrozes diagnoze tiek uzskatīta par iespējamu; ja hlorīdu koncentrācija pārsniedz 100 mmol/l, tā ir droša; šajā gadījumā hlora un nātrija koncentrācijas starpībai nevajadzētu pārsniegt 8–10 mmol/l. Hadsons (1983) iesaka, ja nātrija un hlorīdu saturs sviedros ir uz robežas, veikt prednizolona testu (5 mg iekšķīgi 2 dienas, kam seko elektrolītu noteikšana sviedros). Personām, kuras neslimo ar cistisko fibrozi, nātrija līmenis sviedros samazinās līdz normas apakšējai robežai; cistiskās fibrozes gadījumā tas nemainās. Katram bērnam ar hronisku klepu ieteicams veikt sviedru testu.
Asins pilienu vai DNS paraugu analīze, lai noteiktu cistiskās fibrozes gēna galvenās mutācijas, ir visjutīgākais un specifiskākais diagnostikas tests. Tomēr šī metode ir piemērota tikai valstīm, kurās delta-P508 mutāciju līmenis pārsniedz 80%. Turklāt šī metode ir ļoti dārga un tehniski sarežģīta.
Cistiskās fibrozes pirmsdzemdību diagnostika tiek veikta, nosakot sārmainās fosfatāzes izoenzīmus augļūdeņos. Šī metode kļūst iespējama no 18. līdz 20. grūtniecības nedēļai.
Galvenie cistiskās fibrozes diagnosticēšanas kritēriji ir šādi:
- norādes par aizkavētu fizisko attīstību bērnībā, atkārtotām hroniskām elpceļu slimībām, dispepsijas traucējumiem un caureju, cistiskās fibrozes klātbūtni tuviem radiniekiem anamnēzē;
- hronisks obstruktīvs bronhīts, bieži atkārtojas, ar bronhektāzes un plaušu emfizēmas attīstību, bieži atkārtojas pneimonija;
- hronisks recidivējošs pankreatīts ar ievērojamu eksokrīnās funkcijas samazināšanos, malabsorbcijas sindroms;
- paaugstināts hlora saturs pacienta sviedros;
- neauglība ar saglabātu seksuālo funkciju.
Cistiskās fibrozes veiksmīgu diagnostiku un diferenciāldiagnozi veicina riska grupu identificēšana.
Cistiskās fibrozes skrīninga programma
- Vispārēja asins, urīna, krēpu analīze.
- Krēpu bakterioloģiskā analīze.
- Koproloģiskā analīze.
- Bioķīmiskā asins analīze: kopējā olbaltumvielu un olbaltumvielu frakciju, glikozes, bilirubīna, aminotransferāžu, sārmainās fosfatāzes, gamma-glutamiltranspeptidāžu, kālija, kalcija, dzelzs, lipāzes, amilāzes, tripsīna noteikšana.
- Aizkuņģa dziedzera eksokrīnās funkcijas un zarnu absorbcijas funkcijas izpēte.
- Plaušu fluoroskopija un rentgenogrāfija, plaušu datortomogrāfija.
- EKG.
- Ehokardiogrāfija.
- Bronhoskopija un bronhogrāfija.
- Spirometrija.
- Sviedru tests.
- Konsultācija pie ģenētiķa.
- Asins pilienu vai DNS paraugu analīze, lai noteiktu cistiskās fibrozes gēna galvenās mutācijas.
Kas ir jāpārbauda?
Kādi testi ir vajadzīgi?
Kurš sazināties?
Prognoze
Cistiskās fibrozes pacientu vidējais izdzīvošanas vecums ir no 35 līdz 40 gadiem. Vīriešiem vidējais izdzīvošanas vecums ir augstāks nekā sievietēm.
Ar modernām ārstēšanas stratēģijām 80% pacientu sasniedz pilngadību. Tomēr cistiskā fibroze ievērojami ierobežo pacienta funkcionālās spējas. Šai slimībai joprojām nav izārstēšanas.