Raksta medicīnas eksperts

Jaunas publikācijas
Prioni - prionu slimību izraisītāji
Pēdējā pārskatīšana: 06.07.2025

Visi iLive saturs ir medicīniski pārskatīts vai pārbaudīts, lai nodrošinātu pēc iespējas lielāku faktisko precizitāti.
Mums ir stingras iegādes vadlīnijas un tikai saikne ar cienījamiem mediju portāliem, akadēmiskām pētniecības iestādēm un, ja vien iespējams, medicīniski salīdzinošiem pārskatiem. Ņemiet vērā, ka iekavās ([1], [2] uc) esošie numuri ir klikšķi uz šīm studijām.
Ja uzskatāt, ka kāds no mūsu saturiem ir neprecīzs, novecojis vai citādi apšaubāms, lūdzu, atlasiet to un nospiediet Ctrl + Enter.
Lēnām vīrusu infekcijām raksturīgi īpaši kritēriji:
- neparasti ilgs inkubācijas periods (mēneši, gadi);
- specifisks orgānu un audu, galvenokārt centrālās nervu sistēmas, bojājums;
- lēna, vienmērīga slimības progresēšana;
- neizbēgams letāls iznākums.

Daži patogēni, kas izraisa akūtas vīrusu infekcijas, var izraisīt arī lēnas vīrusu infekcijas. Piemēram, masalu vīruss dažreiz izraisa SSPE, bet masaliņu vīruss izraisa progresējošas iedzimtas masaliņas un masaliņu panencefalītu.
Tipisku lēnu dzīvnieku vīrusu infekciju izraisa visna/madi vīruss, kas ir retrovīruss. Tas ir lēnas vīrusu infekcijas un progresējošas pneimonijas izraisītājs aitām. Smadzeņu baltā viela tiek iznīcināta, attīstās paralīze (visna - novājēšana); rodas hronisks plaušu un liesas iekaisums.
Slimības, kas pēc savām pazīmēm ir līdzīgas lēnajām vīrusu infekcijām, izraisa prioni - prionu infekciju izraisītāji. Prionu slimības ir progresējošu cilvēku un dzīvnieku centrālās nervu sistēmas traucējumu grupa. Cilvēkiem ir traucēta centrālās nervu sistēmas darbība, rodas personības izmaiņas un kustību traucējumi. Slimības simptomi parasti ilgst no vairākiem mēnešiem līdz vairākiem gadiem, beidzoties ar nāvi. Iepriekš prionu infekcijas tika uzskatītas par kopā ar tā sauktajiem lēno vīrusu infekciju izraisītājiem.
Daži prionu slimību izraisītāji vispirms uzkrājas limfoīdos audos. Prioni, nonākot smadzenēs, uzkrājas lielos daudzumos, izraisot amiloidozi (ārpusšūnu disproteinozi, kurai raksturīga amiloīda nogulsnēšanās ar audu atrofijas un sklerozes attīstību) un astrocitozi (astrocītu neirogliju proliferāciju, gliju šķiedru hiperprodukciju). Veidojas fibrili, olbaltumvielu vai amiloīda agregāti un sūkļveida izmaiņas smadzenēs (transmisīvās sūkļveida encefalopātijas). Rezultātā mainās uzvedība, tiek traucēta kustību koordinācija, attīstās izsīkums ar letālu iznākumu. Neveidojas imunitāte. Prionu slimības ir konformācijas slimības, kas attīstās nepareizas šūnu olbaltumvielu, kas nepieciešamas organisma normālai darbībai, locīšanās (pareizas konformācijas pārkāpuma) rezultātā. Prionu pārnešanas ceļi ir dažādi:
- gremošanas ceļš — inficēti dzīvnieku izcelsmes produkti, pārtikas piedevas no neapstrādātiem liellopu orgāniem utt.:
- pārnešana ar asins pārliešanas palīdzību, dzīvnieku izcelsmes zāļu ievadīšana, orgānu un audu transplantācija, inficētu ķirurģisko un zobārstniecības instrumentu lietošana;
- pārnešana ar imunobioloģisko preparātu palīdzību (ir zināma 1500 aitu inficēšanās ar PrP''' ar smadzeņu formalīna vakcīnu no slimām aitām).
Patoloģiskie prioni, nonākot zarnās, tiek transportēti asinīs un limfā. Pēc perifēras replikācijas liesā, apendiksā, mandelēs un citos limfoīdos audos tie caur perifērajiem nerviem tiek pārnesti uz smadzenēm (neiroinvazija). Ir iespējama tieša prionu iekļūšana smadzenēs caur hematoencefālisko barjeru. Iepriekš tika uzskatīts, ka centrālā nervu sistēma ir vienīgie audi, kuros uzkrājas patoloģiskie prioni, taču ir parādījušies pētījumi, kas ir mainījuši šo hipotēzi. Izrādījās, ka prionu uzkrāšanās liesā ir saistīta ar folikulu dendritisko šūnu palielināšanos un darbību.
 [
[ Prionu īpašības
Prionu proteīna normālo šūnu izoformu ar molekulmasu 33–35 kDa nosaka prionu proteīna gēns (prionu gēns — PrNP — atrodas cilvēka 20. hromosomā). Normālais gēns atrodas uz šūnas virsmas (noenkurots membrānā ar molekulas glikoproteīnu), ir jutīgs pret proteāzi. Tas regulē nervu impulsu pārraidi, dienas ciklus, oksidācijas procesus, piedalās vara metabolismā centrālajā nervu sistēmā un kaulu smadzeņu cilmes šūnu dalīšanās regulēšanā. Turklāt prionu gēns ir atrodams liesā, limfmezglos, ādā, kuņģa-zarnu traktā un folikulārajās dendritiskajās šūnās.
Patoloģisko prionu izplatīšanās
Prionu pārveidošanās izmainītās formās notiek, kad tiek izjaukts kinētiski kontrolētais līdzsvars starp tiem. Procesu pastiprina patoloģiskā (PrP) vai eksogēnā priona daudzuma palielināšanās. PrP ir normāls proteīns, kas noenkurots šūnas membrānā. PrP' ir globulārs hidrofobs proteīns, kas veido agregātus ar sevi un PrP'' uz šūnas virsmas: rezultātā PrP' tiek pārveidots par PrP'', un pēc tam cikls turpinās. PrP'' patoloģiskā forma uzkrājas neironos, piešķirot šūnai porainu izskatu.
Kuru
Prionu slimība, kas iepriekš bija izplatīta papuāniešu vidū (tas nozīmē trīci vai drebēšanu) Jaungvinejas salas austrumu daļā. Slimības infekciozās īpašības pierādīja K. Gajduseks. Patogēns tiek pārnests ar pārtiku rituāla kanibālisma rezultātā - ēdot nepietiekami termiski apstrādātas, ar prioniem inficētas mirušo radinieku smadzenes. Centrālās nervu sistēmas bojājumu rezultātā tiek traucētas kustības un gaita, parādās drebuļi un eiforija ("smieklu nāve"). Inkubācijas periods ilgst 5-30 gadus. Pacients mirst pēc gada.
Kreicfelda-Jakoba slimība
Prionu slimība, kas izpaužas kā demence, redzes un smadzenīšu traucējumi un kustību traucējumi ar letālu iznākumu pēc 4–5 mēnešu slimības klasiskajā Kreicfelda-Jakoba slimības variantā un pēc (3–14 mēnešu) jaunajā Kreicfelda-Jakoba slimības variantā. Inkubācijas periods var sasniegt 20 gadus. Iespējami dažādi inficēšanās ceļi un slimības cēloņi:
- lietojot uzturā nepietiekami termiski apstrādātus dzīvnieku izcelsmes produktus, piemēram, gaļu un smadzenes no govīm ar liellopu sūkļveida encefalopātiju;
- audu transplantācijas laikā, piemēram, radzenes transplantācijas, asins pārliešanas, hormonu un citu dzīvnieku izcelsmes bioloģiski aktīvu vielu lietošanas, ketguta lietošanas, piesārņotu vai nepietiekami sterilizētu ķirurģisko instrumentu lietošanas, prosektoriālas manipulācijas;
- PrR hiperprodukcijas un citu stāvokļu gadījumā, kas stimulē PrR' pārvēršanas procesu par PrR".
Slimība var attīstīties arī prionu gēna reģiona mutācijas vai ievietošanas rezultātā. Slimības ģimenes raksturs ir izplatīts ģenētiskas predispozīcijas dēļ uz Kreicfelda-Jakoba slimību. Jaunajā Kreicfelda-Jakoba slimības variantā traucējumi attīstās jaunākā vecumā (vidējais vecums 28 gadi), atšķirībā no klasiskā varianta (vidējais vecums 65 gadi). Jaunajā Kreicfelda-Jakoba slimības variantā patoloģisks prionu proteīns uzkrājas ne tikai centrālajā nervu sistēmā, bet arī limforetikulārajos audos, tostarp mandeles.
Gerstmaņa-Štrauslera-Šeinkera sindroms
Iedzimta prionu slimība, ko pavada demence, hipotonija, rīšanas traucējumi (disfāgija), dizartrija. Bieži vien ir ģimenes raksturs. Inkubācijas periods ir no 5 līdz 30 gadiem. Slimība sākas 50–60 gadu vecumā, tās ilgums ir no 5 līdz 13 gadiem.
Iedzimta letāla bezmiegs
Autoimūna slimība ar progresējošu bezmiegu, simpātisku hiperreaktivitāti (hipertensiju, hipertermiju, hiperhidrozi, tahikardiju), trīci, ataksiju, multiklonālu lēkmi, halucinācijām. Miegs ir smagi traucēts. Nāve iestājas, progresējot sirds un asinsvadu mazspējai.
Skrāpēt
Skrepi (no angļu valodas scrape — nokasīt) ir aitu un kazu prionu slimība (kašķis), kas rodas ar centrālās nervu sistēmas bojājumiem, progresējošiem kustību traucējumiem, stipru ādas niezi (kašķis) un beidzas ar dzīvnieka nāvi.
Govju sūkļveida encefalopātija
Liellopu slimība, kam raksturīgi centrālās nervu sistēmas bojājumi, kustību koordinācijas traucējumi un neizbēgama dzīvnieka nāve. Slimības epidēmija pirmo reizi uzliesmoja Lielbritānijā. Tā bija saistīta ar dzīvnieku barošanu ar gaļas un kaulu miltiem, kas saturēja patoloģiskus prionus. Inkubācijas periods ir no 1,5 līdz 15 gadiem. Visvairāk inficējas dzīvnieku smadzenes, muguras smadzenes un acs āboli.
Prionu slimību laboratoriskā diagnostika
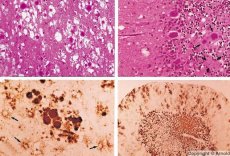
Diagnostikas laikā tiek konstatētas sūkļveida izmaiņas smadzenēs, astrocitoze (glioze) un iekaisuma infiltrātu neesamība. Smadzenes tiek iekrāsotas amiloīda noteikšanai. Prionu smadzeņu slimību olbaltumvielu marķieri tiek atklāti cerebrospinālajā šķidrumā (izmantojot ELISA). Tiek veikta prionu gēna ģenētiskā analīze (PCR).
Prionu slimību profilakse
Instrumentu un vides priekšmetu dekontaminācijai ieteicama autoklāvēšana (134°C temperatūrā 18 min; 121°C temperatūrā 1 h), sadedzināšana, papildu apstrāde ar balinātāju un viennormālu NaCl šķīdumu 1 h. Nespecifiskai profilaksei ir ieviesti ierobežojumi dzīvnieku izcelsmes zāļu lietošanai un ir aizliegta dzīvnieku izcelsmes hipofīzes hormonu ražošana. Ir ierobežota cietā smadzeņu apvalka transplantācija. Strādājot ar pacientu dialoga šķidrumiem, tiek izmantoti gumijas cimdi.