Raksta medicīnas eksperts

Jaunas publikācijas
Keratoderma: cēloņi, simptomi, diagnostika, ārstēšana
Pēdējā pārskatīšana: 23.04.2024

Visi iLive saturs ir medicīniski pārskatīts vai pārbaudīts, lai nodrošinātu pēc iespējas lielāku faktisko precizitāti.
Mums ir stingras iegādes vadlīnijas un tikai saikne ar cienījamiem mediju portāliem, akadēmiskām pētniecības iestādēm un, ja vien iespējams, medicīniski salīdzinošiem pārskatiem. Ņemiet vērā, ka iekavās ([1], [2] uc) esošie numuri ir klikšķi uz šīm studijām.
Ja uzskatāt, ka kāds no mūsu saturiem ir neprecīzs, novecojis vai citādi apšaubāms, lūdzu, atlasiet to un nospiediet Ctrl + Enter.
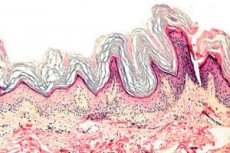
Keratoderma - dermatožu grupa, ko raksturo keratinizācijas procesu pārkāpums, - pārmērīgs ragu veidošanās galvenokārt plaukstām un zolēm.
Slimības cēloņi un patogēze nav pilnībā noskaidrota. Pētījumi ir konstatēts, ka keratoderma, ko izraisa mutācija gēnos, kuri kodē keratīnu 6., 9., 16. In patoģenēzē liela nozīme ir A vitamīna deficīts hormonālā disfunkcija, jo īpaši seksuāla dziedzeri, baktēriju un vīrusu infekcijām. Tie ir viens no iedzimtu slimību simptomiem un iekšējo orgānu audzējiem (parapsoriska keratoderma).
Simptomi. Atšķirt difūza (keratoderma Unna-Toast, keratoderma Meleda, keratoderma Papillons-Lefebvre, mutiliruyushaya keratoderma un sindromi, ieskaitot izkliedētā keratoderma kā viens no galvenajiem simptomiem) un fokusa (jāizplata uznes keratoderma Fischer-Buschke, akrokeratoelastoidoz Bone norobežo keratoderma Bryuaauera-Frantseshesti taisnu keratodermija Fuchs uc) keratoderma.
Wynna-Toast keratoderma (sinonīmi: plaušu un pēdu iedzimta iekaisums, Wyna-Toast sindroms) pārsvarā dominē autosomāli. Tiek atzīmēta plaukstu un pēdu (dažkārt tikai pēdu), kas attīstās pirmajos divos dzīves gados, pārmērīga keratinizācija. Āda un patoloģisks process sākas ar plaukstu un zolīšu ādas nelielu sabiezēšanu, kas veido ādas krāsu ar eritēmu, uz veselīgas ādas robežas. Laika gaitā uz to virsmas parādās gludi, dzeltenīgi ragu slāņi. Bojājums reti nokļūst uz plaukstas vai pirkstu aizmugures virsmas. Dažiem pacientiem var rasties virspusējas vai dziļas plaisas, un ir novērojama vietēja hiperhidroze. Attiecībā uz tēvocīti, ko autors novēroja no mātes puses, brālis un dēls cieta keratodermu no Wyna-Toast.
Ir gadījumi, nagu infekciju (pietūkuma), zobu, matu pie keratoderma Uiny-Toast kombinācijā ar dažādām skeleta anomālijas un traucējumu iekšējo orgānu, nervu un endokrīno sistēmu.
Histopatoloģija. Histoloģiskā izmeklēšana atklāj izteiktu hiperkeratozi, granulozi, acanthozi, mazus iekaisuma infiltrātus dermas augšējā slānī. Diferenciālā diagnoze. Slimība ir jādiferencē no citu veidu keratodermijas.
Keratoderma Meleda (ražotāji: Meleda slimība, iedzimta progresējoša akrokeratoma, plaukstu-pēdu keratozes transgradientny Siemens, iedzimtām, plaukstu-pēdu keratoze progradiently kurš) iedzimta autosomāli recesīvā. Tādā veidā pastāv keratoderma ragveida slāņi ir bieza, dzeltena, brūna līdz tumši plaisas. Bojājuma malās redzams violeti violeta loka platums, kas ir vairāki milimetri. Raksturīgs ir procesa pāreja uz rokas un kāju, apakšdelmu un kāju aizmugurējo virsmu. Lielākajai daļai pacientu ir vietēja hiperhidroze. Šajā sakarā virsma plaukstu un pēdu kļūt nedaudz mitru, un pārklāti ar melniem punktiņiem (cauruļvadi ar sviedru dziedzeru).
Slimība var attīstīties 15-20 gadus. Nagi sabiezē, deformējas.
Histopatoloģija. Histoloģiskā izmeklēšana atklāj hiperkeratozi, reizēm - acanthozi, dermas papilāru slānī - hronisku iekaisuma infiltrāciju.
Diferenciālā diagnoze. Keratoderma Melle jānošķir no keratoderma Unny-Toast.
Keratodermija Papillon-Lefevre (sinonīms: palmāra-plantāra hiperkeretoze ar periodontītu) tiek mantota autosomāli-recesīvi.
Slimība izpaužas 2-3 gadu dzīves laikā. Slimības klīniskais attēlojums ir līdzīgs Melle klīniskajam attēlam. Turklāt raksturīgās izmaiņas zobu (anomāliju izvirduma primāro un pastāvīgo zobu ar kariess, gingivīts, strauji attīstās periodonta slimību ar priekšlaicīgu zaudējumu zobiem).
Histopatoloģija. Histoloģiskā izmeklēšana atklāj dermā visu epidermas, it īpaši ragu, slāņa biezumu - nenozīmīgu limfocītu un himtiocītu šūnu kopu veidošanos.
Diferenciālā diagnoze. Slimība ir jānošķir no citām keratodermijām. Šajā gadījumā svarīga atšķirtspēja ir zobu raksturīgā patoloģija, kas nenotiek citos pārmantotās difūzās keratodermiskās formās.
Mutiliruyuschaya keratoderma (ražotāji: Fonvinkelya sindroms, iedzimts mutiliruyuschaya keratome) - veida difūzu keratoderma, iedzimta autosomāli dominantu. Tas attīstās 2. Dzīves gadā, to raksturo disfunkcionēti ragveida slāņi uz palmu un pēdu ādas ar hiperhidrozi. Laika gaitā uz pirkstiem tiek veidotas auklas veida rievas, kas noved pie kontrakcijas un spontānas pirkstu amputācijas. Folikulāra keratozi izpaužas uz rokas muguras virsmas, kā arī elkoņa un ceļa locītavas zonā. Nagu plati mainītas (bieži vien pēc pulksteņa stikla veida). Aprakstīti hipogonādisma gadījumi, rubīna alopēcija, dzirdes zudums, pachionihija.
Histopatoloģija. Histoloģiskā izmeklēšana atklāj spēcīgu hiperkeratozi, granulozi, acantozi, dermā - nelielus iekaisuma infiltrātus, kas sastāv no limfocītiem un himtiocītiem.
Diferenciālā diagnoze. Diferencējot mutācijas keratodermiju no citām difūzās keratodermijas formām, vispirms jāņem vērā mutācijas efekts, kas nav raksturīgs citām formām. Veicot visu difūzās keratoderma diferenciāldiagnostiku, jāatceras, ka tas var būt viens no vairākiem iedzimtiem sindromiem.
Ārstēšana. Vispārējā keratoderma terapijā parādās neotigazons. Zāles deva ir atkarīga no procesa smaguma un ir 0,3-1 mg / kg pacienta svara. Ja neotigazona nav, ilgstoši ieteikt A vitamīnu devā no 100 līdz 300 000 mg dienā. Ārējā terapija sastāv no ziedēm ar aromātiskiem retinoīdiem, keratolītiskiem un steroīdiem līdzekļiem.
 [
[Kas tevi traucē?
Kas ir jāpārbauda?
Kā pārbaudīt?