Raksta medicīnas eksperts

Jaunas publikācijas
Iedzimts nefrīts (Alporta sindroms) bērniem
Pēdējā pārskatīšana: 05.07.2025

Visi iLive saturs ir medicīniski pārskatīts vai pārbaudīts, lai nodrošinātu pēc iespējas lielāku faktisko precizitāti.
Mums ir stingras iegādes vadlīnijas un tikai saikne ar cienījamiem mediju portāliem, akadēmiskām pētniecības iestādēm un, ja vien iespējams, medicīniski salīdzinošiem pārskatiem. Ņemiet vērā, ka iekavās ([1], [2] uc) esošie numuri ir klikšķi uz šīm studijām.
Ja uzskatāt, ka kāds no mūsu saturiem ir neprecīzs, novecojis vai citādi apšaubāms, lūdzu, atlasiet to un nospiediet Ctrl + Enter.
Iedzimts nefrīts (Alporta sindroms) ir ģenētiski noteikta iedzimta neimūna glomerulopātija, kas izpaužas kā hematūrija (dažreiz ar proteinūriju), progresējoša nieru darbības pasliktināšanās ar hroniskas nieru mazspējas attīstību, bieži vien kombinējot ar sensorineirālu kurlumu un redzes traucējumiem.
Slimību pirmo reizi 1902. gadā aprakstīja L. G. Gutrijs, kurš novēroja ģimeni, kurā hematūrija tika novērota vairākās paaudzēs. 1915. gadā A. F. Hērsts aprakstīja urēmijas attīstību vienas ģimenes locekļiem. 1927. gadā A. Alports pirmo reizi identificēja dzirdes zudumu vairākiem radiniekiem ar hematūriju. 20. gadsimta 50. gados tika aprakstīti acu bojājumi līdzīgas slimības gadījumā. 1972. gadā pacientiem ar iedzimtu hematūriju, veicot nieru audu morfoloģisku pētījumu, Hinglais et al. atklāja nevienmērīgu glomerulu bazālo membrānu paplašināšanos un stratifikāciju. 1985. gadā tika identificēts iedzimta nefrīta ģenētiskais pamats - mutācija IV tipa kolagēna gēnā (Fiengold et al., 1985).
Slimības ģenētiskā rakstura izpēte ļāva secināt, ka iedzimta nefrīta fenotipisko izpausmju atšķirības (ar vai bez dzirdes zuduma) ir saistītas ar mutanta gēna ekspresijas pakāpi. Tādējādi pašlaik visi klīniskie varianti tiek uzskatīti par vienas slimības izpausmēm, un termins "iedzimts nefrīts" ir sinonīms terminam "Alporta sindroms".
Saskaņā ar epidemioloģiskajiem pētījumiem, iedzimts nefrīts rodas ar biežumu 17 uz 100 000 bērniem.
 [
[ Alporta sindroma cēloņi
Slimības ģenētiskais pamats ir IV tipa kolagēna a-5 ķēdes gēna mutācija. Šis tips ir universāls nieru, gliemežnīcas, lēcas kapsulas, tīklenes un radzenes bazālajām membrānām, kas ir pierādīts pētījumos, kuros izmanto monoklonālās antivielas pret šo kolagēna frakciju. Nesen ir norādīta iespēja izmantot DNS zondes iedzimta nefrīta pirmsdzemdību diagnostikai.
Tiek uzsvērta visu ģimenes locekļu testēšanas ar DNS zondēm nozīme, lai identificētu mutanta gēna nesējus, kam ir liela nozīme, veicot medicīnisko un ģenētisko konsultēšanu ģimenēm ar šo slimību. Tomēr līdz pat 20% ģimeņu nav radinieku, kas cieš no nieru slimībām, kas liecina par augstu patoloģiskā gēna spontāno mutāciju biežumu. Lielākajai daļai pacientu ar iedzimtu nefrītu ģimenēs ir cilvēki ar nieru slimībām, dzirdes zudumu un redzes patoloģiju; svarīgas ir asinsradniecības laulības starp cilvēkiem ar vienu vai vairākiem senčiem, jo radniecīgu personu laulībās palielinās varbūtība saņemt tos pašus gēnus no abiem vecākiem. Ir noteikti autosomāli dominējošie, autosomāli recesīvie un dominējošie, ar X hromosomu saistītie pārnešanas ceļi.
Bērniem visbiežāk izšķir trīs iedzimta nefrīta veidus: Alporta sindromu, iedzimtu nefrītu bez dzirdes zuduma un ģimenes labdabīgu hematūriju.
Alporta sindroms ir iedzimts nefrīts ar dzirdes traucējumiem. Tā pamatā ir kombinēts defekts nieru, auss un acu struktūru glomerulu bazālās membrānas kolagēna struktūrā. Klasiskā Alporta sindroma gēns atrodas X hromosomas garās rokas lokusā 21-22q. Vairumā gadījumu tas tiek mantots dominējošā veidā, saistīts ar X hromosomu. Šajā ziņā Alporta sindroms vīriešiem ir smagāks, jo sievietēm mutanta gēna funkciju kompensē vesela otrās, nebojātās hromosomas alēle.
Iedzimta nefrīta attīstības ģenētiskais pamats ir IV tipa kolagēna alfa ķēžu gēnu mutācijas. Ir zināmas sešas IV tipa kolagēna G alfa ķēdes: a5 un a6 ķēžu gēni (Col4A5 un Col4A5) atrodas X hromosomas garajā rokā 21-22q zonā; a3 un a4 ķēžu gēni (Col4A3 un Col4A4) atrodas 2. hromosomā; a1 un a2 ķēžu gēni (Col4A1 un Col4A2) atrodas 13. hromosomā.
Vairumā gadījumu (80–85%) tiek atklāts ar X hromosomu saistīts slimības iedzimtības modelis, kas saistīts ar Col4A5 gēna bojājumiem delēcijas, punktmutāciju vai splaisinga traucējumu rezultātā. Pašlaik ir atrastas vairāk nekā 200 Col4A5 gēna mutācijas, kas ir atbildīgas par IV tipa kolagēna a5 ķēžu sintēzes traucējumiem. Ar šāda veida iedzimtību slimība izpaužas abu dzimumu bērniem, bet zēniem tā ir smagāka.
Mutācijas Col4A3 un Col4A4 gēnu lokos, kas ir atbildīgi par IV tipa kolagēna a3 un a4 ķēžu sintēzi, tiek mantotas autosomāli. Saskaņā ar pētījumiem, autosomāli dominējošais mantojuma veids ir novērots 16% iedzimta nefrīta gadījumu, bet autosomāli recesīvais veids - 6% pacientu. Ir zināmi aptuveni 10 Col4A3 un Col4A4 gēnu mutāciju varianti.
Mutāciju rezultāts ir IV tipa kolagēna montāžas procesu pārkāpums, kas noved pie tā struktūras pārkāpuma. IV tipa kolagēns ir viena no galvenajām glomerulu bazālās membrānas, kohleārā aparāta un acs lēcas sastāvdaļām, kuras patoloģija tiks atklāta iedzimta nefrīta klīnikā.
IV tipa kolagēns, kas ir daļa no glomerulu bazālās membrānas, galvenokārt sastāv no divām a1 ķēdēm (IV) un vienas a2 ķēdes (IV), kā arī satur a3, a4, a5 ķēdes. Visbiežāk X hromosomu saistītā mantojumā Col4A5 gēna mutāciju pavada a3, a4, a5 un a6 ķēžu neesamība IV tipa kolagēna struktūrā, un palielinās o1 un a2 ķēžu skaits glomerulu bazālajā membrānā. Šīs parādības mehānisms nav skaidrs, tiek pieņemts, ka cēlonis ir pēctranskripcijas izmaiņas mRNS.
α3, α4 un α5 ķēžu trūkums glomerulu bazālo membrānu IV tipa kolagēna struktūrā noved pie to retināšanas un trausluma Alporta sindroma sākumposmā, kas klīniski biežāk izpaužas kā hematūrija (retāk kā hematūrija ar proteinūriju vai tikai proteinūriju), dzirdes zudums un lentikonuss. Turpmāka slimības progresēšana noved pie bazālo membrānu sabiezēšanas un caurlaidības traucējumiem slimības vēlīnās stadijās, ar V un VI tipa kolagēna proliferāciju tajās, kas izpaužas kā proteinūrijas palielināšanās un nieru darbības samazināšanās.
Iedzimtā nefrīta pamatā esošās mutācijas raksturs lielā mērā nosaka tā fenotipisko izpausmi. X hromosomas delēcijas gadījumā ar vienlaicīgu Col4A5 un Col4A6 gēnu mutāciju, kas ir atbildīgi par IV tipa kolagēna a5 un a6 ķēžu sintēzi, Alporta sindroms tiek kombinēts ar barības vada un dzimumorgānu leiomiomatozi. Saskaņā ar pētījumu datiem, Col4A5 gēna mutācijas gadījumā, kas saistīta ar delēciju, tiek atzīmēta lielāka patoloģiskā procesa smaguma pakāpe, nieru bojājumu kombinācija ar ekstrarenālām izpausmēm un hroniskas nieru mazspējas agrīna attīstība, salīdzinot ar šī gēna punktveida mutāciju.
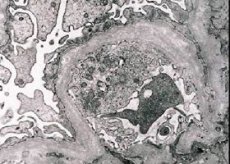
Morfoloģiski elektronmikroskopija atklāj glomerulu bazālo membrānu (īpaši lamina densa) retināšanu un stratifikāciju, kā arī elektronblīvu granulu klātbūtni. Glomerulu bojājumi vienam un tam pašam pacientam var būt heterogēni, sākot no minimāliem fokāliem mezangiāliem bojājumiem līdz glomerulosklerozei. Alporta sindroma glomerulīts vienmēr ir imūnnegatīvs, kas to atšķir no glomerulonefrīta. Raksturīgās pazīmes ir tubulāras atrofijas attīstība, limfohistiocitāra infiltrācija un "putu šūnu" klātbūtne ar lipīdu ieslēgumiem - lipofāgiem. Slimības progresēšanas laikā atklājas glomerulu bazālo membrānu sabiezējums un izteikta destrukcija.
Atklājas noteiktas izmaiņas imūnsistēmā. Pacientiem ar iedzimtu nefrītu ir pazemināts Ig A līmenis un tendence paaugstināt IgM koncentrāciju asinīs, IgG līmenis var būt paaugstināts slimības sākumposmā un samazināties vēlīnā stadijā. Iespējams, IgM un G koncentrācijas paaugstināšanās ir sava veida kompensējoša reakcija, reaģējot uz IgA deficītu.
T-limfocītu sistēmas funkcionālā aktivitāte ir samazināta; tiek atzīmēta selektīva B-limfocītu, kas ir atbildīgi par Ig A sintēzi, samazināšanās, tiek traucēta imunitātes fagocītu saite, galvenokārt neitrofilu ķemotakses un intracelulāro gremošanas procesu traucējumu dēļ.
Izmeklējot nieru biopsiju pacientiem ar Alporta sindromu, elektronmikroskopijas dati atklāj glomerulu bazālās membrānas ultrastrukturālas izmaiņas: retināšanu, struktūras traucējumus un glomerulu bazālo membrānu šķelšanos ar izmaiņām to biezumā un nevienmērīgām kontūrām. Iedzimta nefrīta sākumposmā defekts nosaka glomerulu bazālo membrānu retināšanu un trauslumu.
Glomerulu membrānu retināšanās ir labvēlīgāka pazīme un biežāk sastopama meitenēm. Pastāvīgāka elektronmikroskopiska iedzimta nefrīta pazīme ir bazālās membrānas plīsums, un tās bojāejas smagums korelē ar procesa smagumu.
Alporta sindroma simptomi bērniem
Pirmie Alporta sindroma simptomi izolēta urīnceļu sindroma veidā visbiežāk tiek atklāti bērniem pirmajos trīs dzīves gados. Vairumā gadījumu slimība tiek atklāta nejauši. Urīnceļu sindroms tiek atklāts bērna profilaktiskās apskates laikā, pirms uzņemšanas bērnu aprūpes iestādē vai ARVI laikā. Patoloģijas gadījumā urīnā ARVI laikā. Iedzimta nefrīta gadījumā, atšķirībā no iegūta glomerulonefrīta, latentā perioda nav.
Slimības sākumposmā bērna veselība cieš maz, raksturīga iezīme ir urīnceļu sindroma noturība un rezistence. Viena no galvenajām pazīmēm ir dažādas smaguma pakāpes hematūrija, kas novērota 100% gadījumu. Hematūrijas pakāpes palielināšanās tiek novērota elpceļu infekciju laikā vai pēc tām, fiziskās aktivitātes laikā vai pēc profilaktiskajām vakcinācijām. Proteinūrija vairumā gadījumu nepārsniedz 1 g/dienā, slimības sākumā var būt nepastāvīga, procesam progresējot, proteinūrija palielinās. Periodiski urīna nogulumos var būt leikocitūrija ar limfocītu pārsvaru, kas ir saistīta ar intersticiālu izmaiņu attīstību.
Pēc tam tiek traucēta daļēja nieru darbība, pasliktinās pacienta vispārējais stāvoklis: parādās intoksikācija, muskuļu vājums, arteriāla hipotensija, bieži dzirdes traucējumi (īpaši zēniem) un dažreiz redzes traucējumi. Intoksikācija izpaužas kā bālums, nogurums un galvassāpes. Slimības sākotnējā stadijā dzirdes zudumu vairumā gadījumu atklāj tikai ar audiogrāfiju. Dzirdes zudums Alporta sindromā var rasties dažādos bērnības periodos, bet visbiežāk dzirdes zudums tiek diagnosticēts 6–10 gadu vecumā. Dzirdes zudums bērniem sākas ar augstām frekvencēm, sasniedzot ievērojamu pakāpi gaisa un kaulu vadītspējā, pārejot no skaņu vadoša uz skaņu uztverošu dzirdes zudumu. Dzirdes zudums var būt viens no pirmajiem slimības simptomiem un var būt pirms urīnceļu sindroma.
20% gadījumu pacientiem ar Alporta sindromu ir izmaiņas redzes orgānos. Visbiežāk konstatētās anomālijas ir lēcas anomālijas: sferofokija, priekšējais, aizmugurējais vai jauktais lēcas acs iekšpuse un dažādas kataraktas. Ģimenēs ar Alporta sindromu ir ievērojama miopijas biežums. Vairāki pētnieki šajās ģimenēs pastāvīgi atzīmē divpusējas perimakulāras izmaiņas spilgti bālganas vai dzeltenīgas granulācijas dzeltenajā ķermenī. Viņi uzskata šo pazīmi par pastāvīgu simptomu ar augstu diagnostisko vērtību Alporta sindroma gadījumā. K. S. Čugs un līdzautori (1993) oftalmoloģiskā pētījumā pacientiem ar Alporta sindromu konstatēja redzes asuma samazināšanos 66,7% gadījumu, priekšējo lēcas acs iekšpusi 37,8% gadījumu, tīklenes plankumus 22,2% gadījumu, kataraktu 20% un keratokonusu 6,7% gadījumu.
Dažiem bērniem ar iedzimtu nefrītu, īpaši, ja attīstās nieru mazspēja, tiek atzīmēta ievērojama fiziskās attīstības atpalicība. Progresējot nieru mazspējai, attīstās arteriāla hipertensija. Bērniem to biežāk atklāj pusaudža gados un vecākās vecuma grupās.
Pacientiem ar iedzimtu nefrītu raksturīgas dažādas (vairāk nekā 5–7) saistaudu dismorfogenēzes stigmas. Starp saistaudu stigmām pacientiem visbiežāk sastopamas acu hipertelorisms, augstas aukslējas, sakodiena anomālijas, anomāla ausu priekškambaru forma, mazā pirkstiņa izliekums uz rokām un "sandales sprauga" uz pēdām. Iedzimtam nefrītam raksturīga dismorfogenēzes stigmu vienveidība ģimenē, kā arī to bieža izplatība starp probandu radiniekiem, pa kuru līniju slimība tiek pārnesta.
Slimības agrīnajās stadijās tiek konstatēta izolēta daļēju nieru funkciju samazināšanās: aminoskābju, elektrolītu transportēšana, koncentrācijas funkcija, acidoģenēze, vēlākas izmaiņas ietekmē gan nefrona proksimālās, gan distālās daļas funkcionālo stāvokli un tām raksturīgi kombinēti daļēji traucējumi. Glomerulārās filtrācijas samazināšanās notiek vēlāk, biežāk pusaudža gados. Progresējot iedzimtam nefrītam, attīstās anēmija.
Tādējādi iedzimtu nefrītu raksturo pakāpeniska slimības gaita: vispirms latentā stadija vai slēpti klīniskie simptomi, kas izpaužas ar minimālām izmaiņām urīnceļu sindromā, tad pakāpeniska procesa dekompensācija notiek ar nieru darbības samazināšanos ar izteiktiem klīniskiem simptomiem (intoksikācija, astēnija, attīstības aizkavēšanās, anēmija). Klīniskie simptomi parasti parādās neatkarīgi no iekaisuma reakcijas slāņojuma.
Iedzimts nefrīts var izpausties dažādos vecuma periodos, kas ir atkarīgs no gēna darbības, kas atrodas represētā stāvoklī līdz noteiktam laikam.
Klasifikācija
Pastāv trīs iedzimta nefrīta veidi
- I variants - klīniski izpaužas kā nefrīts ar hematūriju, dzirdes zudumu un acu bojājumiem. Nefrīta gaita ir progresējoša, attīstoties hroniskai nieru mazspējai. Iedzimtības veids ir dominējošs, saistīts ar X hromosomu. Morfoloģiski tiek atklāts bazālās membrānas struktūras pārkāpums, tās retināšana un šķelšanās.
- II variants - klīniski izpaužas kā nefrīts ar hematūriju bez dzirdes zuduma. Nefrīta gaita ir progresējoša, attīstoties hroniskai nieru mazspējai. Iedzimtības veids ir dominējošs, saistīts ar X hromosomu. Morfoloģiski tiek konstatēta glomerulu kapilāru bazālās membrānas (īpaši laminadensas) retināšanās.
- III variants - labdabīga ģimenes hematūrija. Gaita ir labvēlīga, hroniska nieru mazspēja neattīstās. Mantojuma veids ir autosomāli dominējošais vai autosomāli recesīvais. Ar autosomāli recesīvo mantojuma veidu sievietēm tiek novērota smagāka slimības gaita.
Alporta sindroma diagnoze
Tiek ierosināti šādi kritēriji:
- vismaz divu pacientu ar nefropātiju klātbūtne katrā ģimenē;
- hematūrija kā galvenais nefropātijas simptoms probandā;
- dzirdes zuduma klātbūtne vismaz vienam ģimenes loceklim;
- hroniskas nieru mazspējas attīstība vienam vai vairākiem radiniekiem.
Dažādu iedzimtu un iedzimtu slimību diagnostikā liela vieta tiek piešķirta visaptverošai pieejai izmeklēšanai un, galvenais, uzmanības pievēršanai datiem, kas iegūti, sastādot bērna ciltsrakstu. Alporta sindroma diagnoze tiek uzskatīta par pamatotu gadījumos, kad pacientam tiek konstatētas 3 no 4 tipiskām pazīmēm: hematūrija un hroniska nieru mazspēja ģimenē, neirosensoriska dzirdes zuduma klātbūtne, redzes patoloģija pacientam, glomerulu bazālās membrānas šķelšanās pazīmju noteikšana ar izmaiņām tās biezumā un nelīdzenām kontūrām biopsijas elektronmikroskopiskajā izmeklējumā.
Pacienta izmeklēšanai jāietver klīniskās un ģenētiskās izpētes metodes; mērķtiecīga slimības vēstures izpēte; vispārēja pacienta izmeklēšana, ņemot vērā diagnostiski nozīmīgus kritērijus. Kompensācijas stadijā patoloģiju var atklāt, tikai koncentrējoties uz tādiem sindromiem kā iedzimta slodze, hipotensija, vairākas disembriogenēzes stigmas, izmaiņas urīnceļu sindromā. Dekompensācijas stadijā var parādīties ekstrarenāli simptomi, piemēram, smaga intoksikācija, astēnija, aizkavēta fiziskā attīstība, anēmija, kas izpaužas un pastiprinās, pakāpeniski samazinoties nieru darbībai. Lielākajai daļai pacientu, samazinoties nieru darbībai, novēro: samazināta acido- un aminoģenēze; 50% pacientu atzīmē ievērojamu nieru sekrēcijas funkcijas samazināšanos; ierobežotas urīna optiskā blīvuma svārstības; filtrācijas ritma traucējumus un pēc tam glomerulārās filtrācijas samazināšanos. Hroniskas nieru mazspējas stadija tiek diagnosticēta, ja pacientiem 3-6 mēnešus vai ilgāk ir paaugstināts urīnvielas līmenis asins serumā (vairāk nekā 0,35 g/l), un glomerulārās filtrācijas samazināšanās līdz 25% no normas.
Iedzimta nefrīta diferenciāldiagnostika galvenokārt jāveic ar iegūtā glomerulonefrīta hematūrisko formu. Iegūtajam glomerulonefrītam visbiežāk ir akūta sākšanās, 2–3 nedēļu periods pēc infekcijas, ekstrarenālas pazīmes, tostarp hipertensija jau no pirmajām dienām (iedzimta nefrīta gadījumā, gluži pretēji, hipotensija), samazināta glomerulārā filtrācija slimības sākumā, nav daļēju tubulāro funkciju traucējumu, savukārt iedzimta nefrīta gadījumā tās ir klātesošas. Iegūts glomerulonefrīts rodas ar izteiktāku hematūriju un proteinūriju, ar paaugstinātu ESR. Diagnostiska vērtība ir tipiskām izmaiņām glomerulu bazālajā membrānā, kas raksturīgas iedzimtam nefritam.
Diferenciāldiagnostika no dismetaboliskās nefropātijas tiek veikta ar hronisku nieru mazspēju, ģimenē klīniski atklātas heterogēnas nieru slimības, un var būt nefropātijas spektrs no pielonefrīta līdz urolitiāzei. Bērniem bieži ir sūdzības par sāpēm vēderā un periodiski urinēšanas laikā, urīna nogulsnēs - oksalāti.
Ja ir aizdomas par iedzimtu nefrītu, pacients jānosūta uz specializētu nefroloģijas nodaļu, lai precizētu diagnozi.
Kas ir jāpārbauda?
Kā pārbaudīt?
Kādi testi ir vajadzīgi?
Kurš sazināties?
Prognoze
Iedzimta nefrīta prognoze vienmēr ir nopietna.
Prognoziski nelabvēlīgi iedzimta nefrīta gaitas kritēriji ir:
- vīriešu dzimums;
- hroniskas nieru mazspējas agrīna attīstība ģimenes locekļiem;
- proteinūrija (vairāk nekā 1 g dienā);
- glomerulu bazālo membrānu sabiezēšana saskaņā ar mikroskopiju;
- akustiskais neirīts;
- delēcija Col4A5 gēnā.
Labdabīgas ģimenes hematūrijas prognoze ir labvēlīgāka.
Использованная литература