Raksta medicīnas eksperts

Jaunas publikācijas
Tricher Collins sindroms
Pēdējā pārskatīšana: 23.04.2024

Visi iLive saturs ir medicīniski pārskatīts vai pārbaudīts, lai nodrošinātu pēc iespējas lielāku faktisko precizitāti.
Mums ir stingras iegādes vadlīnijas un tikai saikne ar cienījamiem mediju portāliem, akadēmiskām pētniecības iestādēm un, ja vien iespējams, medicīniski salīdzinošiem pārskatiem. Ņemiet vērā, ka iekavās ([1], [2] uc) esošie numuri ir klikšķi uz šīm studijām.
Ja uzskatāt, ka kāds no mūsu saturiem ir neprecīzs, novecojis vai citādi apšaubāms, lūdzu, atlasiet to un nospiediet Ctrl + Enter.

Kad augļa kaulu attīstības traucējumi procesi ir smaga kraniofaciāliem deformāciju, un viens no šķirnēm šādu patoloģiju ir Treacher Collins sindroms (TCS) vai mandibulofastsialny, t.i., sejas-žokļu dysostosis.
Slimības kods saskaņā ar ICD 10: XVII klase (iedzimtas anomālijas, deformācijas un hromosomu patoloģijas), Q75.4 - mandibolofaciālā disostoze.
Cēloņi no Tricher Collins sindroma
Šis sindroms tika nosaukts pēc izcilā britu oftalmologa Edvarda Tricera Kolinsa, kurš aprakstīja patoloģijas galvenās iezīmes vairāk nekā simts gadus atpakaļ. Tomēr Eiropas ārsti bieži sauc šāda veida anomālijām sejas kaulu un žokļu slimību vai sindromu Franceschetti - balstoties uz plašu izpēti par Šveices oftalmologa Adolf Franceschetti, kurš izdomāts termins "mandibulofastsialny dysostosis" vidū pagājušā gadsimta. Medicīnas aprindās nosaukums tiek lietots arī - Franceschetti-Kolinsa sindroms.
Izraisa Treacher Collins sindroms - gēna mutācijas TCOF1 (locus 5q31.3-33.3 hromosomas), kas kodē nucleolar fosfoproteīna, kas ir atbildīgs par veidošanās kraniofaciāliem daļu no cilvēka embrija. Šīs olbaltuma daudzuma priekšlaicīga samazināšanās rezultātā tiek pārtraukta biogēna un rRNS funkcija. Saskaņā ar ģenētiķiem Cilvēka Genoma izpētes programmas, šie procesi izraisīt samazinājumu izplatīšanu embrija nervu šķautnes šūnām - veltņa gar nervu grāvī, kas izstrādes gaitā embrija stāšanās neironu caurules aizveras.
Veidošanās no priekšējās daļas galvaskausa audu notiek sakarā ar transformācijas un šūnu diferenciācijas augšējo (galva) daļa neironu crest ka migrē gar neironu cauruli uz pirmo un otro branchial arkas embriju. Un šo šūnu deficīts izraisa kraniofakālās deformācijas. Anomāliju sastopamības kritiskais periods ir no 18 līdz 28 dienām pēc apaugļošanas. Pabeidzot migrācijas neironu šķautnes šūnu (ceturtajā nedēļu grūtniecības) ir izveidotas gandrīz visu brīvs mezenhimâlo audu sejas, kas vēlāk (5 līdz 8 nedēļas) atšķirt uz skeleta un saistaudu audiem daļās sejas, kakla, rīkles, ausu (ieskaitot iekšējie) un nākotnes zobi.
Pathogenesis
Par Treacher Collins sindroma patoģenēzes bieži darbināt ģimenēs, un anomāliju ir mantotas autosomāli dominējošā principa, lai gan ir gadījumi, autosomāli recesīvo pārvades defektiem (mutācijas citu gēnu, jo īpaši, POLR1C un POLR1D). Visvairāk neprognozējams zarnu un sejas folikulāro disostozi ir tas, ka bērnus mutācijas pārmanto tikai 40-48% gadījumu. Tas nozīmē, ka 52-60% no pacientiem izraisa Treacher Collins sindromu nav saistīts ar klātbūtni anomāliju ģimenē, un tiek uzskatīts, ka patoloģiju rodas sporādiski ģenētiskas mutācijas de novo. Visticamāk, jaunās mutācijas atspoguļo teratogēnas ietekmes ietekmi uz augli grūtniecības laikā.
Starp cēloņiem šī sindroma teratogēna lietprateji lielas devas etanola (etilspirts), radiācija, cigarešu dūmiem, tsitomegavirus un toksoplazmozes, kā arī par herbicīdu glifosātu (Raundal, Glifor TORNADO, un citi.). Un jatrogēno faktoru saraksts ietvēra preparātus pūtītēm un seboreju ar 13-cis-retinoīdu skābi (izotretinoīns, akutaāns); Antikonvulsējošs līdzeklis Fenitoīns (Dilantīns, Epanupīns); psihotropās zāles, diazepāms, valium, relanijs, seduksens.
Simptomi no Tricher Collins sindroma
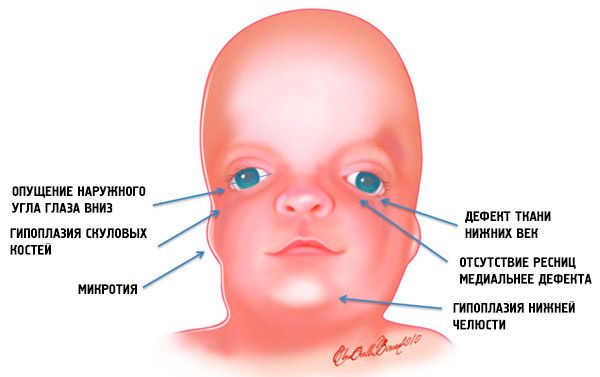
Lielākoties mandibolofosciālās disostozes klīniskās pazīmes un to smaguma pakāpe ir atkarīga no gēnu mutāciju izpausmju iezīmēm. Un pirmās šīs anomālijas pazīmes lielākajā daļā gadījumu ir redzamas bērnam tūlīt pēc viņa dzimšanas: sejai ar Tricher Collins sindromu ir raksturīgs izskats. Turklāt morfoloģiskās anomālijas parasti ir divpusējas un simetriskas.
Visredzamākie Tricera Kolinsa sindroma simptomi ir:
- mazattīstība (hipoplāzija) sejas kauli galvaskausa: zygomatic, zygomatic process no pieres kaulu, sānu pterygoid plāksnes, blakusdobumu, apakšžokļa kaulu epifīzes un izvirzījumiem (condyles);
- apakšējā žokļa kaulu (mikrogānijas) kaulu nepietiekama attīstība un biezāka nekā parasti apakšējā leņķa;
- degunam ir normāls lielums, taču tas šķiet lieliski, pateicoties psihiatrisko arku hipoplāzijai un tempļu apgabala nepietiekamam attīstījumam vai zigomitāru arku trūkumam;
- acs šķēli tiek dilstoši, tas ir, acu iegriezums ir patoloģisks, ārējie stūri noliecami uz leju;
- apakšējo plakstiņu defekti (koloboma) un daļēja skropstu trūkums uz tiem;
- neregulāras formas ausis ar lielu noviržu diapazonu, līdz to atrašanās vietai apakšējās žokļa stūrī, cilpiņu trūkums, aklās fistulas starp kazas ausu un mutes stūri utt .;
- ārējās dzirdes kanāla sašaurināšanās vai infekcija (atrezija) un vidusauss sēžakaru anomālijas;
- nepakļauta vai slikta dūša;
- gremošanas traucējumi (rētas un elpošanas ceļu sašaurināšanās);
- cietās aukslējas (vilka mutē) nemierināšanās, kā arī mīkstajām ausīm trūkums, saīsināšana vai nekustīgums.
Šādām anatomiskām patoloģijām visos gadījumos ir komplikācijas. Tie ir funkcionāli dzirdes traucējumi vadītāja (vadītāja) dzirdes zuduma vai pilnīgas kurluma formā; redzes traucējumi nepareizas acs ābolu formas dēļ; Alauces defekti rada grūtības barojot un norijot. Ar zobiem saistīti ar zobiem sakropļojumi (nepareiza ēsma), kas savukārt rada problēmas ar košļājamo un artikulāciju. Mutes dobuma patoloģijas izskaidro deguna balsi.
Komplikācijas un sekas
Par kraniofaciāliem novirzēm Treacher Collins sindromu sekas izpaužas ar to, ka pēc piedzimšanas viņa normāli intelektuālo spēju, bet gan tāpēc, ka dzirdes defektus un citi traucējumi atzīmēta sekundāro garīgo atpalicību.
Turklāt bērni ar šādiem defektiem akūtiski apzinās savu zemestību un cieš, kas negatīvi ietekmē viņu nervu sistēmu un psihi.
Diagnostika no Tricher Collins sindroma
Tricher Kolinsa sindroma postnatālā diagnoze galvenokārt balstās uz klīniskajām pazīmēm. Maxillofacial dysostosis ir viegli noteikt, kad pilnībā ekspresivitāte sindroms, bet, kad ir minimāli izteikta slimības simptomi, ar pareizu diagnozi var būt problēmas.
Šajā gadījumā īpaša uzmanība ir jāpievērš, lai novērtētu visas ar anomālijām saistītās funkcijas, jo īpaši tās, kas ietekmē elpošanu (miega apnojas draudi). Tiek veikta hemoglobīna barošanas un piesātinājuma efektivitātes novērtēšana un kontrole ar skābekli.
Nākotnē - 5.-6. Dienā pēc dzemdībām - ir nepieciešams noskaidrot dzirdes bojājuma pakāpi ar audioloģiskās pārbaudes palīdzību, kas jāveic dzemdību nama slimnīcā.
Tiek veikta pārbaude, kuras laikā instrumentālo diagnostiku veic ar galvaskausa-sejas disormorfoloģijas fluoroskopiju; pantomogrāfija (sejas locekļa kaulu struktūru panorāmas rentgenogramma); pilna kraniālā datortomogrāfija dažādās izstādēs; DNS vai smadzeņu MRI, lai noteiktu iekšējā dzirdes aparāta stāvokli.
Agrākais - Pirmsdzemdību - diagnoze kraniofaciāliem novirzēm klātbūtnē Treacher Collins sindromu, ģimenes vēsture ir iespējams, horiona villus paraugu ņemšanas laikā 10-11 grūtniecības nedēļās (procedūra draud aborts un infekcijas dzemdē).
Notiek arī ģimenes locekļu asins analīzes; 16-17 grūtniecības nedēļās tiek veikta amnija šķidruma (transabdominālais amniocentēzes) analīze; 18-20 grūtniecības nedēļās tiek veikta fetoskopija, un asinis ņem no placentas augļu traukiem.
Bet visbiežāk šā sindroma prenatālajā diagnozē auglim tiek izmantota ultraskaņa (20-24 grūtniecības nedēļās).
Kādi testi ir vajadzīgi?
Diferenciālā diagnoze
Šie paši metodes speciālisti izmantot, kad nepieciešams diferenciāldiagnostiku, atpazīt klusi izrunāts Treacher Collins sindromu un atšķirt to no citiem iedzimtas anomālijas kraniofaciāliem kauliem, jo īpaši: sindromi Apert, Crouzon, Nagera Peters-Hevelsa, Hellerman-Staefa, kā arī ar hemifacial microsomia (Goldenhar sindroms), hypertelorism, agri imperforate galvaskausa šuves (craniostenosis) vai pārkāpjot izkausētas sejas kaulu (kraniostenoze).
Prognoze
Ko var prognozēt šī patoloģija? Tas ir atkarīgs no deformācijas pakāpes un simptomu intensitātes. Tricker Collins sindroms ir mūža diagnoze.
 [25]
[25]