Raksta medicīnas eksperts

Jaunas publikācijas
Polijas sindroms
Pēdējā pārskatīšana: 23.04.2024

Visi iLive saturs ir medicīniski pārskatīts vai pārbaudīts, lai nodrošinātu pēc iespējas lielāku faktisko precizitāti.
Mums ir stingras iegādes vadlīnijas un tikai saikne ar cienījamiem mediju portāliem, akadēmiskām pētniecības iestādēm un, ja vien iespējams, medicīniski salīdzinošiem pārskatiem. Ņemiet vērā, ka iekavās ([1], [2] uc) esošie numuri ir klikšķi uz šīm studijām.
Ja uzskatāt, ka kāds no mūsu saturiem ir neprecīzs, novecojis vai citādi apšaubāms, lūdzu, atlasiet to un nospiediet Ctrl + Enter.
Reta anomālija ķermeņa struktūras intrauterīnā veidošanās procesā, kas galvenokārt sastāv no krūšu kaula hipoplāzijas un liela krūšu muskuļa ribas daļas vai tās pilnīgas neesamības. Viņš nēsā angļu ķirurgu, kurš studentu gados aprakstīja ekstremitāšu un muskuļu defektu, kad viņš strādāja morgā. A.Polande nebija pirmais, pirms viņiem atsevišķos gadījumos XIX gadsimta sākumā jau pievērsa uzmanību Francijā un Vācijā, bet tā bija viņa publikācija, kas uzsāka nopietnu šīs iedzimtas patoloģijas izpēti. 20. Gadsimta mijā J. Thompsons publicēja slimības pilnu aprakstu. Kopš tā laika aptuveni 500 šādu gadījumu ir aprakstīti pasaules medicīnas literatūrā.
 [
[Epidemioloģija
Saslimstību statistika liecina, ka dzimstības mala muskuļu patoloģijas, izteikti dažādās pakāpēs, ir vidēji viens jaundzimušo no 30000, vai nedaudz vairāk, nekā dzīvi dzimušajiem. Biežāk ar šādām anomālijām rodas zēni.
Līdz 80% no deformācijas Polijas sindromā ir taisnās puses. Traucējumi ir izteikti dažādās pakāpēs, un nav konstatēta atbilstība starp smaguma pakāpes novirzes no formas krūtīm un rokām.
Cēloņi polijas sindroms
Bērnu piedzimšanas iemesli ar šo anomāliju joprojām ir hipotētiski. Mantojuma veids un gēns, kas pārraida šo patoloģiju, nav definēts, bet Polijas sindroms raksturo retu ģimenes vēsturi. Tas uzņem recesīvu mantojumu. Tiek uzskatīts, ka varbūtība slimības pārnēsāšanai no slimiem vecākiem uz viņu bērniem ir aptuveni 50%. Lielākā daļa gadījumu ir vieni. Bērnu ar šo anomāliju dzimšanas riska faktori ir ārēji un iekšēji teratogēni ietekme uz embriju orgānu un sistēmu dēšanas un attīstības laikā. Ir vairākas hipotēzes, kas izskaidro šīs ribu muskuļu defekta etioloģiju un patoģenēzi, bet neviens no tiem nav pilnībā apstiprināts. Visticamākais pieņēmums ir tas, ka jebkurš nelabvēlīgs faktors izraisa embriju asins piegādes trūkumu grūtniecības sestā nedēļā, kad veidojas subklāvija artērija. Tas izraisa tā nepietiekamu attīstību (lūmena sašaurināšanos) un nepietiekamu asins piegādi, kas izraisa mīksto audu un kaulu vietēju hipoplaziju. Bojājuma apmēru nosaka pēc artērijas un / vai tā filiāļu bojājuma pakāpes.
Starp iemesliem minēti arī embriju krūšu kurvju-muskuļu audu šūnu migrācijas vai intrauterīnās traumas migrācijas pārkāpumi. Bet pietiekami ticamu pierādījumu līdz šim nav nevienas no šīm hipotēzēm.
Simptomi polijas sindroms
Šīs iedzimtas anomālijas pirmās pazīmes ir redzamas jau pēc zīdaiņa, pateicoties raksturīgajam krūšu muskuļu un asiņainas izskata. Un roku hipoplāzijas klātbūtnē - no dzimšanas.
Sindroma simptomu komplekss ir šāds:
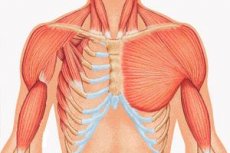
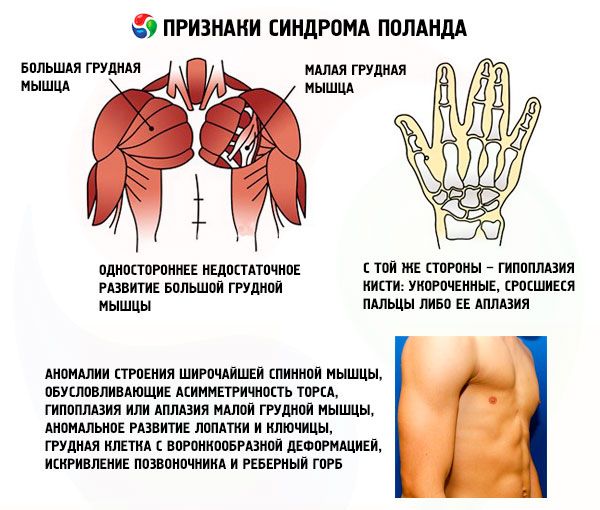
- liela krūšu muskuļa vai tās fragmentu vienpusēja nepietiekama attīstība, visbiežāk - krūšu kauls un kaklā;
- tajā pašā pusē - rokas hipoplāzija: saīsināts, kausētais pirksts vai tā aplasija; nepietiekama krūts attīstība vai tā trūkums, atelija; zemādas tauku slāņa izdalījumi; asiņainu matiņu trūkums; anomālijas skrimšļa / kaulu kāju audu struktūrā vai to pilnīga neesamība (parasti III un IV).
Otrajā daļā aprakstīto funkciju klātbūtne nav obligāta, tos var apvienot ar pirmo, izmantojot dažādas iespējas.
Bez šiem, ļoti reti var būt anomālijas struktūru Lat, nosacījumu par asimetrijas rumpja, hipoplāziju vai aplāzija no pectoralis nepilngadīgais muskuļu, patoloģiska lāpstiņas un atslēgas kaula attīstību, krūtīm ar piltuves formas deformācijas, mugurkaula deformācija un ribu kupris.
Kad kreisā defekta puse bieži tiek novērota transponējot iekšējos orgānus, jo īpaši sirds tiek pārvietota pa labi. Normālā sirds atrašanās vietā kopā ar ribu trūkumu tā praktiski nav aizsargāta un tās palpēšana ir pamanāma zem ādas.
Polijas sindroms bērniem parasti ir ievērojams no dzimšanas brīža, bet dažos gadījumos nelieli defekti tiek konstatēti aptuveni trīs gadus.
Pēc lokalizācijas, krūškurvja struktūras elementu defekti ir sadalīti priekšējās, pakaļējās un sānu sienu deformācijās.
Polijas sindroms meitenēm pubertātē pat pat mazākā mērā izjūt tam, ka bojātā pusē esošā krūtiņa nav veidojusies vai atpaliek attīstībā un atrodas daudz augstāka nekā ar parasto. Vieglos zēnu saslimšanas gadī- jumos sindroms dažreiz tiek novērots diezgan novēloti pusaudža gados, kad nav iespējams izsūknēt muskuļus no defektīvās puses.
Polijas sindroms sievietēm neietekmē hormonālo fonu un spēju iedzīt bērnu.
Vairumā gadījumu Polijas sindroms ir kosmētiskais defekts: krūškurvja muskuļi visbiežāk ir deformēti vai nav, nav krūškurvja defekta, un ir pilnvērtīga suka. Aizsargā augšējo ekstremitāšu mehāniskās funkcijas un nekas neļauj šiem pacientiem intensīvi iesaistīties sportā.
Tomēr ir arī citi, vairāk traumatiskie šīs patoloģijas veidi. Šādu lietu sekas un sarežģījumi ir nedaudz nopietnāki. Atkarībā no deformācijas smaguma pakāpes, pacientam var rasties elpošanas funkcijas traucējumi un hemodinamika. Gadījumā, ja nepastāv kastrētu krampju rāmis, bieži tiek konstatēta plaušu trūce, un elpošanas traucējumi rodas no paša dzimšanas brīža.
Retos gadījumos kreisās puses patoloģijas kopā ar ribu trūkumu ar normālu orgānu izvietojumu sirds ir tieši zem ādas. Šāda pacienta dzīvi pastāvīgi apdraud briesmas, kas saistītas ar iespējamu traumu un sirdsdarbības apstāšanos.
Bērnam ar smagu krūšu kurvja defektu parasti ir ar hemodinamiku saistītas problēmas sakarā ar samazinātu sistolisko un paaugstināto diastolisko artēriju kombinācijā ar paaugstinātu venozo spiedienu. Šiem bērniem raksturīgs paaugstināts nogurums, astēniskais sindroms, viņi var atpaliek no saviem vienaudžiem fiziskajā attīstībā.
Polijas sindroma izpausmes skar gan subklāvijas artērijas struktūru, gan tās filiāles, kas rada apstākļus arteriālās asinsrites traucējumiem defekta pusē.
Tiek novērotas dažas anatomiskas novirzes no svarīgāko iekšējo orgānu struktūras un izvietojuma. To smaguma pakāpe var ievērojami sarežģīt pacienta stāvokli. Šī sirds novirze no normālas atrašanās vietas vienā virzienā vai otrā virzienā, līdz pat transponēšanai, tās robežu paplašināšanai vai pagriešanās pulksteņrādītāja virzienā, plaušu un nieru hipoplazija defektīvā pusē.
Posmi
Šajā slimībā krūšu kurvja veidošanos veido četri posmi.
Pirmais ir raksturīgs lielākajai daļai zināmu gadījumu, kad tikai mīkstie audi ir pārmērīgi attīstīti, un krūšu kurvja forma un riņķa skrimšļa un kaulozes daļa ir normāla.
Otrais - ja pieskārās deformācija krūšu kurvis: bojāts pusē, kad uzglabā kaulains un skrimšļveida porcijas ribām nedaudz nomākts ar piekrastes skrimšļos, polubokom izvietoti kaula un pretējā pusē novērota bieži apkalpo (keeled) uz krūtīm.
Trešajā posmā struktūra tiek saglabāta kaula daļas ribu, un krimšļa neattīstīta, Krūškurvja ir asimetriska, konusveida uz krūšu kaula celma, bet tika konstatēti bruto anomālijas.
Ceturto posmu raksturo tas, ka no viena līdz četriem (no III līdz VI) nav riņķa kakla un kaulozas daļas. No defektīvās puses trūkst ribu vietā - dobumi, krūšu kauls ir ievērojami izliekts.
Taču jebkurā posmā veidošanās struktūras elementu krūšu kurvis Valsts bērna organisms var būt normāli (kompensēta), ar periodiskiem uzlabojumiem (subkompensēta) un pasliktināšanās ar pieaugumu iekšējo orgānu un skeleta sistēmas (dekompensētas). Tas ir atkarīgs no ķermeņa individuālajām īpašībām, attīstības pakāpes, saslimstības un dzīvesveida.
Diagnostika polijas sindroms
Kolektīvās muskuļu iedzimto patoloģiju nosaka vizuāli, ārsts palpina pacientu un nosaka radiogrāfiju. Parasti tas ir pietiekami, lai atklātu krūšu kurpes apjomu un veidu. Precīzāku slimības attēlu var dot ar datoru un magnētiskās rezonanses attēlveidošanu.
Apaklāvja artērijas ultrasonogrāfija tiek noteikta arī, lai noteiktu tās diametru, smadzeņu ultrasonogrāfisko izmeklēšanu un citu instrumentālo diagnostiku atbilstoši indikācijām.
Lai novērtētu ar to saistītos anatomiskos defektus, konsultācijas ar kardiologu un elektrokardiogrāfiju, nepieciešamas sirds ultraskaņas izmeklēšana, veloergometrija, ehokardiogrāfija un doplerogrāfija.
Elpošanas distressā gadījumā ir nepieciešama pulmonologa konsultācija, kas var noteikt plaušu funkcionālā stāvokļa izpēti, piemēram, spirogrāfiju.
Analīze ar šo slimību parasti ir normas robežās, ja tajā nav vienlaicīgu patoloģiju.
Rūpīgi diagnosticējoši pasākumi ļauj precīzi novērtēt rekonstruktīvo iejaukšanās apjomu.
Kādi testi ir vajadzīgi?
Kurš sazināties?
Prognoze
Vairumā gadījumu iedzimto defektu struktūras organismā tiek samazināta līdz mazattīstītam muskuļu, kas neietekmē darbību ekstremitāšu un iekšējo orgānu, auglības sievietēm, un ir vienreizējās kosmētikas defekts. Pat ar sarežģītākiem kombinētiem bojājumiem, savlaicīga ārstēšana nodrošina pacientam iespēju pilnvērtīgi dzīvot.