Raksta medicīnas eksperts

Jaunas publikācijas
Polimiozīts un dermatomiozīts: cēloņi, simptomi, diagnostika, ārstēšana
Pēdējā pārskatīšana: 05.07.2025

Visi iLive saturs ir medicīniski pārskatīts vai pārbaudīts, lai nodrošinātu pēc iespējas lielāku faktisko precizitāti.
Mums ir stingras iegādes vadlīnijas un tikai saikne ar cienījamiem mediju portāliem, akadēmiskām pētniecības iestādēm un, ja vien iespējams, medicīniski salīdzinošiem pārskatiem. Ņemiet vērā, ka iekavās ([1], [2] uc) esošie numuri ir klikšķi uz šīm studijām.
Ja uzskatāt, ka kāds no mūsu saturiem ir neprecīzs, novecojis vai citādi apšaubāms, lūdzu, atlasiet to un nospiediet Ctrl + Enter.
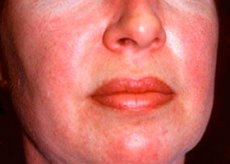
Polimiozīts un dermatomiozīts ir retas sistēmiskas reimatiskas slimības, kam raksturīgas iekaisīgas un deģeneratīvas izmaiņas muskuļos (polimiozīts) vai muskuļos un ādā (dermatomiozīts). Visspecifiskākā ādas izpausme ir heliotropiski izsitumi.
Muskuļu iesaistīšanās ir simetriska un ietver vājumu, nelielu jutīgumu un sekojošu proksimālās iegurņa joslas muskuļu atrofiju. Komplikācijas var ietvert viscerālu iesaistīšanos un ļaundabīgus audzējus. Diagnoze balstās uz klīnisko ainu un muskuļu disfunkcijas novērtējumu, mērot enzīmu līmeni, veicot MRI, elektromiogrāfiju un muskuļu biopsiju. Ārstēšana ietver glikokortikoīdus, dažreiz kombinācijā ar imūnsupresantiem vai intravenoziem imūnglobulīniem.
Sievietes slimo divreiz biežāk nekā vīrieši. Slimība var rasties jebkurā vecumā, bet visbiežāk tā tiek atklāta vecumā no 40 līdz 60 gadiem; bērniem - no 5 līdz 15 gadiem.
 [
[ Kas izraisa dermatomiozītu un polimiozītu?
Tiek uzskatīts, ka slimības cēlonis ir autoimūna reakcija uz muskuļu audiem ģenētiski predisponētiem indivīdiem. Slimība biežāk rodas, ja ir apgrūtināta ģimenes anamnēze un noteiktu HLA antigēnu (DR3, DR52, DR56) nesēji. Iespējamie izraisītāji ir vīrusu miozīts un ļaundabīgi audzēji. Ir ziņojumi par pikornavīrusiem līdzīgu struktūru atklāšanu muskuļu šūnās; turklāt vīrusi var izraisīt līdzīgas slimības dzīvniekiem. Ļaundabīgo audzēju saistība ar dermatomiozītu (daudz retāk nekā ar polimiozītu) liecina, ka audzēja augšana var būt arī slimības attīstības izraisītājs, jo rodas autoimūnas reakcijas uz audzēja un muskuļu audu kopīgiem antigēniem.
IgM, IgG un komplementa trešās komponentes nogulsnes ir atrodamas skeleta muskuļu asinsvadu sieniņās; tas ir īpaši raksturīgi dermatomiozītam bērniem. Pacientiem ar polimiozītu var attīstīties arī citi autoimūni procesi.
Dermatomiozīta un polimiozīta patofizioloģija
Patoloģiskas izmaiņas ietver šūnu bojājumus un atrofiju uz dažādas smaguma pakāpes iekaisuma fona. Augšējo un apakšējo ekstremitāšu muskuļi, kā arī sejas muskuļi tiek bojāti mazākā mērā nekā citi skeleta muskuļi. Rīkles un barības vada augšdaļas viscerālo muskuļu, retāk sirds, kuņģa vai zarnu, bojājumi var izraisīt iepriekš minēto orgānu darbības traucējumus. Augsta mioglobīna koncentrācija rabdomiolīzes dēļ var izraisīt nieru bojājumus. Var rasties arī iekaisuma izmaiņas locītavās un plaušās, īpaši pacientiem, kuriem ir antisintetāzes antivielas.
Dermatomiozīta un polimiozīta simptomi
Polimiozīta sākums var būt akūts (īpaši bērniem) vai subakūts (biežāk pieaugušajiem). Akūta vīrusu infekcija dažreiz notiek pirms slimības izpausmes vai ir tās izraisītājs, kuras visbiežāk sastopamās izpausmes ir proksimālo muskuļu vājums vai izsitumi uz ādas. Sāpes ir izteiktas mazākā mērā nekā vājums. Var attīstīties poliartralģija, Reino sindroms, disfāgija, plaušu darbības traucējumi, vispārēji simptomi (paaugstināta ķermeņa temperatūra, samazināts ķermeņa svars, vājums). Reino sindroms bieži tiek konstatēts pacientiem ar vienlaicīgām saistaudu slimībām.
Muskuļu vājums var progresēt vairāku nedēļu vai mēnešu laikā. Tomēr, lai klīniski izpaustos muskuļu vājums, jābūt skartiem vismaz 50% muskuļu šķiedru (tādēļ muskuļu vājuma klātbūtne norāda uz miozīta progresēšanu). Pacientiem var būt grūtības pacelt rokas virs plecu līmeņa, kāpt pa kāpnēm vai piecelties no sēdus stāvokļa. Smaga iegurņa un plecu joslas muskuļu vājuma dēļ pacienti var būt piesaistīti ratiņkrēslam vai gultai. Ja ir skarti kakla locītāji, galvu nav iespējams pacelt no spilvena. Rīkles un barības vada augšdaļas muskuļu bojājums izraisa rīšanas traucējumus un regurgitāciju. Apakšējo un augšējo ekstremitāšu, kā arī sejas muskuļi parasti netiek skarti. Tomēr var attīstīties ekstremitāšu kontraktūras.
Dermatomiozīta gadījumā novērotie ādas izsitumi parasti ir tumšas krāsas un eritēmatozi. Raksturīga ir arī periorbitāla tūska purpursarkanā krāsā (heliotropiski izsitumi). Ādas izsitumi var būt nedaudz pacelti virs ādas līmeņa un būt gludi vai zvīņaini; izsitumu lokalizācija ir piere, kakls, pleci, krūtis, mugura, apakšdelmi, apakšstilbu apakšējā daļa, uzacis, ceļgalu apvidus, mediālie malleoli, starpfalangu un metakarpofalangeālo locītavu mugurējās virsmas, laterālā pusē (Gotrona simptoms). Iespējama nagu pamatnes vai perifērijas hiperēmija. Uz pirkstu laterālās virsmas ādas var attīstīties deskvamatīvais dermatīts, ko pavada plaisas. Primārie ādas bojājumi bieži izzūd bez sekām, bet var izraisīt sekundāras izmaiņas, piemēram, tumšu pigmentāciju, atrofiju, rētas vai vitiligo. Var attīstīties zemādas kalcifikācijas, īpaši bērniem.
Aptuveni 30% pacientu attīstās poliartralģija vai poliartrīts, ko bieži pavada tūska un locītavu izsvīdums. Tomēr locītavu izpausmju smagums ir neliels. Tās rodas biežāk, ja pacientiem tiek atklātas antivielas pret Jo-1 vai citām sintetāzēm.
Iekšējo orgānu (izņemot rīkli un barības vada augšējo daļu) iesaistīšanās polimiozīta gadījumā ir retāk sastopama nekā citu reimatisku slimību (īpaši SLE un sistēmiskās sklerozes) gadījumā. Reti, īpaši antisintetāzes sindroma gadījumā, slimība izpaužas kā intersticiāls pneimonīts (aizdusas un klepus veidā). Var attīstīties sirds aritmijas un vadīšanas traucējumi, taču tie parasti ir asimptomātiski. Kuņģa-zarnu trakta izpausmes biežāk rodas bērniem, kuriem ir arī vaskulīts, un tās var ietvert vemšanu ar asinīm, melēnu un zarnu perforāciju.
Kur tas sāp?
Polimiozīta klasifikācija
Ir 5 polimiozīta veidi.
- Primārais idiopātiskais polimiozīts, kas var rasties jebkurā vecumā, neietver ādu.
- Primārais idiopātiskais dermatomiozīts ir līdzīgs primārajam idiopātiskajam polimiozītam, bet tas skar ādu.
- Ar ļaundabīgiem audzējiem saistīts polimiozīts un dermatomiozīts var rasties jebkura vecuma pacientiem; to attīstība visbiežāk novērojama gados vecākiem pacientiem, kā arī pacientiem ar citām saistaudu slimībām. Ļaundabīgo audzēju attīstību var novērot gan 2 gadu laikā pirms, gan 2 gadu laikā pēc miozīta sākuma.
- Bērnu polimiozīts vai dermatomiozīts ir saistīts ar sistēmisku vaskulītu.
- Polimiozīts un dermatomiozīts var rasties arī pacientiem ar citām saistaudu slimībām, visbiežāk progresējošu sistēmisku sklerozi, jauktu saistaudu slimību un SLE.
Stumbra muskuļu miozīta iekļaušana polimiozīta grupā ir nepareiza, jo pēdējais ir atsevišķa slimība, kurai raksturīgas līdzīgas klīniskās izpausmes kā hroniskam idiopātiskam polimiozītam. Tomēr tas attīstās vecumā, bieži skar ķermeņa distālo daļu muskuļus (piemēram, augšējo un apakšējo ekstremitāšu), ir ilgāks ilgums, sliktāk reaģē uz ārstēšanu un tam ir raksturīga tipiska histoloģiska aina.
Dermatomiozīta un polimiozīta diagnoze
Polimiozīts ir aizdomas par pacientiem ar sūdzībām par proksimālo muskuļu vājumu ar vai bez jutīguma. Dermatomiozīta izmeklēšana ir nepieciešama pacientiem ar sūdzībām par izsitumiem, kas atgādina heliotropu vai Gotrona zīmi, kā arī pacientiem ar polimiozīta izpausmēm, kas apvienotas ar jebkādiem ādas bojājumiem, kas atbilst dermatomiozītam. Polimiozīta un dermatomiozīta klīniskās izpausmes var līdzināties sistēmiskās sklerozes vai, retāk, SLE vai vaskulīta pazīmēm. Diagnozes noteiktību palielina atbilstība pēc iespējas vairāk no šiem pieciem kritērijiem:
- proksimālo muskuļu vājums;
- raksturīgi ādas izsitumi;
- paaugstināta muskuļu audu enzīmu aktivitāte (kreatīnkināze vai, ja tās aktivitāte nav palielinājusies, aminotransferāzes vai aldolāze);
- raksturīgas izmaiņas miogrāfijā vai MRI;
- raksturīgas histoloģiskas izmaiņas muskuļu audu biopsijā (absolūtais kritērijs).
Muskuļu biopsija var izslēgt dažus klīniski līdzīgus stāvokļus, piemēram, stumbra muskuļu miozītu un rabdomiolīzi vīrusu infekcijas dēļ. Histoloģiskā izmeklēšanā atklātās izmaiņas var atšķirties, taču tipiski ir hronisks iekaisums, muskuļu deģenerācijas un reģenerācijas perēkļi. Pirms potenciāli toksiskas ārstēšanas uzsākšanas ir nepieciešama precīza diagnoze (parasti ar histoloģisku verifikāciju). MRI var noteikt tūskas un iekaisuma perēkļus muskuļos, kam seko mērķtiecīga biopsija.
Laboratoriskie izmeklējumi var apstiprināt vai, gluži pretēji, izslēgt aizdomas par slimības klātbūtni, kā arī ir noderīgi tās smaguma novērtēšanai, kombinācijas iespējamībai ar citām līdzīgām patoloģijām un komplikāciju diagnosticēšanai. Neskatoties uz to, ka dažiem pacientiem tiek atklātas antinukleārās antivielas, šī parādība ir raksturīgāka citām saistaudu slimībām. Apmēram 60% pacientu ir antivielas pret kodolu (PM-1) vai veselu aizkrūtes dziedzera šūnu antigēnu un Jo-1. Autoantivielu loma slimības patogenezē joprojām nav skaidra, lai gan ir zināms, ka antivielas pret Jo-1 ir specifisks antisintetāzes sindroma, tostarp fibrozējošā alveolīta, plaušu fibrozes, artrīta un Reino fenomena, marķieris.
Periodiska kreatīnkināzes aktivitātes novērtēšana ir noderīga ārstēšanas uzraudzībai. Tomēr pacientiem ar smagu muskuļu masas zudumu enzīmu aktivitāte var būt normāla, neskatoties uz hroniska aktīva miozīta klātbūtni. MRI, muskuļu biopsija vai paaugstināta kreatīnkināzes aktivitāte bieži vien ir noderīga, lai diferencētu polimiozīta recidīvu no glikokortikoīdu izraisītas miopātijas.
Tā kā daudziem pacientiem ir nediagnosticēti ļaundabīgi audzēji, daži autori iesaka skrīningu veikt visiem pieaugušajiem ar dermatomiozītu un tiem, kuriem ir polimiozīts un kuri vecāki par 60 gadiem, izmantojot šādu grafiku: fiziskā apskate, tostarp krūšu, iegurņa un taisnās zarnas izmeklēšana (ieskaitot fēču testēšanu uz slēptām asinīm); pilna asins aina; asins ķīmija; mamogrāfija; karcinoembrionālā antigēna tests; urīna analīze; krūškurvja rentgenogrāfija. Daži autori apšauba šādas skrīninga nepieciešamību jaunākiem pacientiem, kuriem nav klīnisku ļaundabīgo audzēju pazīmju.
Kas ir jāpārbauda?
Kā pārbaudīt?