Raksta medicīnas eksperts

Jaunas publikācijas
Martina-Bella sindroms
Pēdējā pārskatīšana: 04.07.2025

Visi iLive saturs ir medicīniski pārskatīts vai pārbaudīts, lai nodrošinātu pēc iespējas lielāku faktisko precizitāti.
Mums ir stingras iegādes vadlīnijas un tikai saikne ar cienījamiem mediju portāliem, akadēmiskām pētniecības iestādēm un, ja vien iespējams, medicīniski salīdzinošiem pārskatiem. Ņemiet vērā, ka iekavās ([1], [2] uc) esošie numuri ir klikšķi uz šīm studijām.
Ja uzskatāt, ka kāds no mūsu saturiem ir neprecīzs, novecojis vai citādi apšaubāms, lūdzu, atlasiet to un nospiediet Ctrl + Enter.
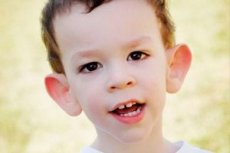
Mārtina-Bella sindromu 1943. gadā aprakstīja ārsti, kuru vārdā tas tika nosaukts. Slimība ir ģenētiska slimība, kas sastāv no garīgas atpalicības. 1969. gadā tika identificētas šai slimībai raksturīgās X hromosomas izmaiņas (trauslums distālajā rokā). 1991. gadā zinātnieki atklāja gēnu, kas ir atbildīgs par šīs slimības attīstību. Šo slimību sauc arī par "trauslā X sindromu". Gan zēni, gan meitenes ir uzņēmīgi pret šo slimību, bet zēni tiek skarti biežāk (3 reizes).
Epidemioloģija
Martina-Bella sindroms ir diezgan izplatīta slimība: no šīs slimības cieš 0,3–1,0 no 1000 vīriešiem un 0,2–0,6 no 1000 sievietēm. Turklāt bērni ar Martina-Bella sindromu piedzimst visos kontinentos ar vienādu biežumu. Acīmredzot tautība, ādas krāsa, acu forma, dzīves apstākļi un cilvēku labklājība neietekmē slimības rašanos. Tās rašanās biežums ir salīdzināms tikai ar Dauna sindroma biežumu (1 slimība no 600–800 jaundzimušajiem). Piektā daļa vīriešu, kas nes izmainīto gēnu, ir veseli, bez klīniskām vai gēnu anomālijām, pārējiem ir garīgās atpalicības pazīmes no vieglas līdz smagai formai. Starp sievietēm nesējām ir slimas nedaudz vairāk nekā trešdaļa.
Traušā X sindroms skar aptuveni 1 no 2500–4000 vīriešiem un 1 no 7000–8000 sievietēm. Nesēju izplatība sieviešu vidū tiek lēsta 1 no 130–250; nesēju izplatība vīriešu vidū tiek lēsta 1 no 250–800.
Cēloņi Martina-Bella sindroms
Martina-Bella sindroms attīstās sakarā ar pilnīgu vai daļēju organisma specifiskas olbaltumvielas ražošanas pārtraukšanu. Tas notiek X hromosomā lokalizētā FMR1 gēna atbildes trūkuma dēļ. Mutācija rodas gēna pārstrukturēšanas rezultātā no nestabiliem gēnu stāvokļu (alēļu) strukturāliem variantiem, nevis no paša sākuma. Slimība tiek pārnesta tikai pa vīriešu līniju, un vīrietis ne vienmēr var būt slims. Vīrieši nesēji nodod gēnu savām meitām nemainītā veidā, tāpēc viņu garīgā atpalicība nav acīmredzama. Turpinot gēna pārnešanu no mātes bērniem, gēns mutē, un parādās visas šai slimībai raksturīgās pazīmes.
Pathogenesis
Martina-Bella sindroma patoģenēze balstās uz gēnu aparāta mutācijām, kas noved pie FMR proteīna, organismam, īpaši neironiem, vitāli svarīga proteīna, kas atrodas dažādos audos, ražošanas bloķēšanas. Pētījumi liecina, ka FMR proteīni ir tieši iesaistīti smadzeņu audos notiekošo translāciju regulēšanas procesos. Šī proteīna trūkums vai tā ierobežota ražošana organismā noved pie garīgas atpalicības.
Slimības patogenezē gēnu hipermetilācija tiek uzskatīta par galveno traucējumu, taču vēl nav bijis iespējams precīzi noteikt šī traucējuma attīstības mehānismu.
Vienlaikus tika atklāta arī patoloģijas lokusa heterogenitāte, kas saistīta ar polialēlismu, kā arī polilokusu. Tika noteikta slimības attīstības alēļu variantu klātbūtne, ko izraisa punktmutāciju esamība, kā arī FMRL tipa gēna destrukcija.
Pacientiem ir arī 2 trausli tripleti, kas ir jutīgi pret folskābi, kas atrodas 300 kb, kā arī 1,5–2 miljoni bp no trauslā tripleta, kas satur FMR1 gēnu. Mutāciju mehānisms FRAXE un FRAXF gēnos (tie ir identificēti iepriekš minētajos trauslajos tripletos) ir saistīts ar traucējumu mehānismu Martina Bella sindromā. Šo mehānismu izraisa GCC un CGG atkārtojumu izplatīšanās, kas izraisa tā saukto CpG salu metilēšanu. Papildus klasiskajai patoloģijas formai pastāv arī 2 reti veidi, kas atšķiras trinukleotīdu atkārtojumu paplašināšanās dēļ (vīriešu un sieviešu mejozes gadījumā).
Tika konstatēts, ka sindroma klasiskajā formā pacientam trūkst īpaša FMR1 tipa nukleocitoplazmatiskā proteīna, kas veic dažādu mRNS saistīšanas funkciju. Turklāt šis proteīns veicina kompleksa veidošanos, kas palīdz veikt translācijas procesus ribosomās.
Simptomi Martina-Bella sindroms
Kā atpazīt slimību bērniem? Kādas ir pirmās pazīmes? Pirmajos bērna dzīves mēnešos Martina-Bella simptomu nav iespējams atpazīt, izņemot to, ka dažreiz tiek novērota muskuļu tonusa samazināšanās. Pēc gada slimības klīniskā aina ir izteiktāka: bērns sāk vēlu staigāt un runāt, dažreiz runas vispār nav. Viņš ir hiperaktīvs, haotiski vicina rokas, baidās no pūļiem un trokšņa, ir spītīgs, ir asi dusmu uzliesmojumi, emocionāla nestabilitāte, rodas epilepsijas lēkmes, neveido acu kontaktu. Pacientiem ar Martina-Bella sindromu slimību nodod arī izskats: ausis ir izvirzītas un lielas, piere ir smaga, seja ir iegarena, zods ir izvirzīts, šķielēšana, platas rokas un kājas. Tiem raksturīgi arī endokrīnās sistēmas traucējumi: bieži liels svars, aptaukošanās, lieli sēklinieki vīriešiem, agrīna pubertāte.

Pacientiem ar Martina-Bella sindromu intelekta līmenis ir ļoti atšķirīgs: no vieglas garīgas atpalicības līdz smagiem gadījumiem. Ja normālam cilvēkam intelekta koeficients (IQ) ir vidēji 100, bet ģēnijam - 130, tad cilvēkiem, kuri ir uzņēmīgi pret šo slimību, tas ir 35-70.
Visus patoloģijas klīniskos simptomus var raksturot ar galveno izpausmju triādi:
- oligofrēnija (IQ ir 35–50);
- dismorfofobija (tiek novērotas izvirzītas ausis un prognātisms);
- makroorhidisms, kas parādās pēc pubertātes sākuma.
Aptuveni 80% pacientu ir arī bikuspidālā vārstuļa prolapss.
Tomēr pilna sindroma forma izpaužas tikai 60% no visiem pacientiem. 10% gadījumu tiek konstatēta tikai garīga atpalicība, bet pārējos gadījumos slimība attīstās ar atšķirīgu simptomu kombināciju.
Starp pirmajām slimības pazīmēm, kas parādās agrīnā vecumā:
- slimajam bērnam ir ievērojama garīga atpalicība salīdzinājumā ar citu vienaudžu attīstību;
- uzmanības un koncentrēšanās traucējumi;
- spēcīga spītība;
- bērni sāk staigāt un runāt diezgan vēlu;
- tiek novērota hiperaktivitāte un runas attīstības traucējumi;
- ļoti spēcīgi un nekontrolējami dusmu lēkmes;
- var attīstīties mutisms - tas ir pilnīgs runas trūkums bērnam;
- mazulis izjūt sociālo trauksmi un spēj krist panikā skaļa trokšņa vai citu skaļu skaņu dēļ;
- bērns nekontrolējami un haotiski vicina rokas;
- tiek novērota kautrība, bērns baidās atrasties pārpildītās vietās;
- dažādu obsesīvu ideju rašanās, nestabils emocionālais stāvoklis;
- Bērns var nevēlēties veidot acu kontaktu ar cilvēkiem.
Pieaugušajiem novēro šādus patoloģijas simptomus:
- specifisks izskats: iegarena seja ar smagu pieri, lielām izvirzītām ausīm, stipri izvirzītu zodu;
- plakanās pēdas, otitis un šķielēšana;
- pubertāte notiek diezgan agri;
- var attīstīties aptaukošanās;
- Diezgan bieži sirds defekti tiek novēroti Martin-Bell sindromā;
- vīriešiem novēro sēklinieku palielināšanos;
- locītavu locītavas kļūst ļoti kustīgas;
- svars un augums strauji palielinās.
Diagnostika Martina-Bella sindroms
Lai diagnosticētu Martina Bella sindromu, jums jāsazinās ar kvalificētu ģenētiķi. Diagnoze tiek noteikta pēc specifiskiem ģenētiskiem testiem, kas ļauj identificēt bojāto hromosomu.
Testi
Slimības agrīnā stadijā tiek izmantota citogēnētiskā metode, kurā no pacienta tiek ņemts šūnu materiāla fragments, kuram pēc tam pievieno folskābi, lai izraisītu izmaiņas hromosomās. Pēc noteikta laika perioda tiek identificēta hromosomas zona, kurā ir manāma retināšana – tā ir trauslā X sindroma klātbūtnes pazīme.
Tomēr šis tests nav piemērots diagnozei slimības vēlākajās stadijās, jo tā precizitāti samazina plaši izplatītā folskābi saturošu multivitamīnu lietošana.
Martina-Bella sindroma integrētā diagnostika ir molekulārā ģenētiskā izmeklēšana, kas sastāv no tā saukto trinukleotīdu atkārtojumu skaita noteikšanas gēnā.
 [ 25 ], [ 26 ], [ 27 ], [ 28 ], [ 29 ]
[ 25 ], [ 26 ], [ 27 ], [ 28 ], [ 29 ]
Instrumentālā diagnostika
Ļoti specifiska instrumentālās diagnostikas metode ir PCR (polimerāzes ķēdes reakcija), kas ļauj pētīt X hromosomā esošo aminoskābju atlikumu struktūru un tādējādi noteikt Martin Bell sindroma klātbūtni.
Pastāv arī atsevišķa, vēl specifiskāka patoloģijas diagnostikas metode – PCR un kapilārās elektroforēzes noteikšanas kombinācija. Šī metode ir ļoti precīza un atklāj hromosomu patoloģiju pacientiem ar primāru olnīcu mazspēju, kā arī ataksijas sindromu.
Defekta klātbūtni var noteikt pēc diagnostikas veikšanas EEG. Pacientiem ar šo slimību ir līdzīga bioelektriskā smadzeņu aktivitāte.
Kādi testi ir vajadzīgi?
Diferenciālā diagnoze
Diferencētas metodes, kas palīdz aizdomām par sindromu, ir šādas:
- klīniski - 97,5% pacientu ir acīmredzamas garīgās atpalicības pazīmes (vidēji smagas vai smagas); 62% ir izvirzītas lielas ausis; 68,4% ir liels izvirzīts zods un piere; 68,4% zēnu ir palielināti sēklinieki, 41,4% ir runas īpatnības (nevienmērīgs runas ātrums, nekontrolējams skaļums utt.);
- citogēns - asinis tiek pārbaudītas limfocītu kultūrai, tiek noteikts šūnu skaits ar trauslu X hromosomu uz 100 pētītajām šūnām;
- Elektroencefalogrāfija - tiek reģistrētas smadzeņu elektrisko impulsu izmaiņas, kas raksturīgas Martina-Bella sindromam.
Kurš sazināties?
Profilakse
Vienīgā slimības profilakses metode ir grūtnieču pirmsdzemdību skrīnings. Ir īpašas pārbaudes, kas ļauj laikus atklāt patoloģiju, pēc kuras ieteicams pārtraukt grūtniecību. Kā alternatīva tiek izmantota IVF, kas var palīdzēt bērnam mantot veselīgu X hromosomu.
Pacienta profilakse ir atkarīga no tā, vai gēnu mutācija ir radusies atkārtoti vai ir iedzimta. Šim nolūkam tiek veikta molekulārā ģenētiskā diagnostika. Fakts, ka tests neatklāja radinieku "trauslo X hromosomu", liecina par labu mutācijas "svaigumam", kas nozīmē, ka bērna ar Martina-Bella sindromu risks ir ļoti mazs. Ģimenēs, kurās ir slimi cilvēki, tests palīdzēs izvairīties no atkārtotiem gadījumiem.
Prognoze
Martina-Bella sindroma prognoze ir labvēlīga dzīvībai, bet ne atveseļošanai. Dzīves ilgums ir atkarīgs no slimības smaguma pakāpes un ar to saistītajiem defektiem. Pacients var dzīvot normālu dzīvi. Smagās Martina-Bella sindroma formās pacientiem draud mūža invaliditāte.
Dzīves ilgums
Martina Bella sindromam nav nopietnas negatīvas ietekmes uz veselību, tāpēc vairuma cilvēku, kuriem diagnosticēta šī patoloģija, paredzamais dzīves ilgums neatšķiras no standarta rādītājiem.