Raksta medicīnas eksperts

Jaunas publikācijas
Alkaptonūrija ir iedzimta enzīmu anomālija.
Pēdējā pārskatīšana: 04.07.2025

Visi iLive saturs ir medicīniski pārskatīts vai pārbaudīts, lai nodrošinātu pēc iespējas lielāku faktisko precizitāti.
Mums ir stingras iegādes vadlīnijas un tikai saikne ar cienījamiem mediju portāliem, akadēmiskām pētniecības iestādēm un, ja vien iespējams, medicīniski salīdzinošiem pārskatiem. Ņemiet vērā, ka iekavās ([1], [2] uc) esošie numuri ir klikšķi uz šīm studijām.
Ja uzskatāt, ka kāds no mūsu saturiem ir neprecīzs, novecojis vai citādi apšaubāms, lūdzu, atlasiet to un nospiediet Ctrl + Enter.
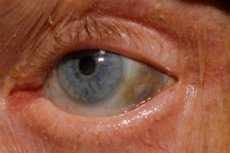
Viens no ļoti retajiem vielmaiņas traucējumiem, alkaptonūrija, attiecas uz iedzimtām anomālijām aminoskābes tirozīna metabolismā.
Šo sindromu var saukt arī par homogentizātoksidāzes deficītu, homogentisinūriju, iedzimtu ohronozi vai melnā urīna slimību.[ 1 ]
Epidemioloģija
Saskaņā ar statistiku, alkaptonūrijas gadījumu skaits nepārsniedz deviņus uz 1 miljonu cilvēku. Un lielākajā daļā Eiropas valstu ir viens gadījums uz 100–250 tūkstošiem jaundzimušo.
Starp Eiropas valstīm izņēmums ir Slovākija (īpaši relatīvi nelielais ziemeļrietumu reģions), kur alkaptonūrijas izplatība ir viens gadījums uz 19 000 jaundzimušajiem. Tas, visticamāk, ir saistīts ar faktu, ka tur dzīvojošo Slovākijas romu ģimeņu vidū radinieku radniecības (laulību starp brālēniem un māsīcām) līmenis ir augstākais Eiropā: 10–14 %. [ 2 ]
Cēloņi alkaptonūrija
Ir noskaidroti precīzi alkaptonūrijas cēloņi kā iedzimts aromātiskās (homocikliskās) α-aminoskābes tirozīna katabolisma (metabolisma noārdīšanās) traucējums: šāda veida vielmaiņas traucējumi ir viena no tūkstošiem gēnu 3. hromosomā homozigotu vai saliktu heterozigotu mutāciju sekas, precīzāk, HGD gēna 3q21-q23 lokusā uz hromosomas garās rokas. Šis gēns kodē aknu enzīma homogentizāt-1,2-dioksigenāzes [ 3 ] (saukta arī par homogentizātskābes oksidāzi vai homogentizātoksidāzi) nukleotīdu sekvences - dzelzi saturošu metaloproteīnu, kas nepieciešams vienam no tirozīna noārdīšanās posmiem organismā [ 4 ], [ 5 ].
Tādējādi alkaptonūrija ir enzīma homogentizāta-1,2-dioksigenāzes defekts vai, precīzāk, tā ģenētiski noteikta deficīta vai pilnīgas neesamības rezultāts. [ 6 ]
Tā kā alkaptonūrija ir iedzimts enzīmu deficīts, tā tiek mantota kā autosomāli recesīva pazīme, tas ir, lai alkaptonūrija rastos bērniem, abiem vecākiem ir jābūt modificētam enzīma gēnam, jo katrs no viņiem nodod bērnam tikai vienu gēna kopiju no divām pieejamajām.
Saskaņā ar jaunākajiem datiem, ir vairāk nekā divi simti HGD gēna modifikācijas variantu, un visbiežāk tiek novērotas missense mutācijas, translokācija un splicēšana.
Riska faktori
Vienīgais šīs iedzimtās enzimopātijas attīstības riska faktors ir tās klātbūtne ģimenes anamnēzē un divu modificētu HGD gēna kopiju mantošana, ja vecākiem nav alkaptonūrijas (anomālijas pārnešanas risks ir 25%) vai vienam no vecākiem ir šī slimība. [ 7 ]
Pathogenesis
Tirozīnam ir galvenā loma olbaltumvielu sintēzē, hromoproteīnu – ādas pigmenta melanīna –, kā arī vairogdziedzera hormonu un kateholamīnu neirotransmiteru ražošanā.
Šūnās esošā tirozīna daudzuma regulēšanas mehānisms ir ļoti sarežģīts, un organisms normalizē tā lieko saturu, to sadalot. Tirozīna katabolisma process, tāpat kā visām aromātiskajām aminoskābēm, ir daudzpakāpju un notiek vairākos posmos. Katrs tirozīna metabolisma sadalīšanās posms notiek, piedaloties noteiktam enzīmam un veidojot starpprodukta savienojumu.
Tātad, vispirms aminoskābe tiek sadalīta līdz para-hidroksifenilpiruvātam, kas tiek pārvērsts par alkaptonu – 2,5-dihidroksifeniletiķskābi jeb homogentisīnskābi. Pēc tam alkaptonam vajadzētu pārvērsties par maletiķskābi, bet tas nenotiek. [ 8 ]
Un alkaptonūrijas patoģenēze sastāv no tirozīna katabolisma bioķīmisko reakciju pārtraukšanas homogentisīnskābes veidošanās stadijā: vienkārši nav nepieciešams enzīms, kas to noārdītu – homogentizātoksidāze.
Homogentisīnskābe organismā netiek izmantota un var uzkrāties, izdaloties caur nierēm. Turklāt tā oksidējas par benzohinoacetātu (benzohinonetiķskābi), kas, saistoties ar audu un ķermeņa šķidrumu molekulām, veido biopolimēru savienojumus, kuru krāsa ir līdzīga melanīnam.
Šo starpproduktu uzkrāšanās audos noved pie skrimšļa audu kolagēna struktūras traucējumiem, kas samazina to elastību – ar daudzu alkaptonūrijas klīnisko pazīmju parādīšanos un komplikāciju attīstību.
Simptomi alkaptonūrija
Alkaptonūrijai jaundzimušajiem un zīdaiņiem raksturīga urīna tumšāka krāsa. Saskaroties ar gaisu, urīns uz autiņbiksītēm, autiņbiksītēm un apakšveļas kļūst tumši brūns; tas ir saistīts ar homogentisīnskābes uzkrāšanos un izdalīšanos, kas oksidējas par benzohinoacetātu. [ 9 ]
Ja nav citu simptomu, alkaptonūrija maziem bērniem bieži netiek laikus atpazīta, jo urīns var kļūt tumšs pēc vairākām urinēšanas stundām. Saskaņā ar dažiem datiem, klīniskajos apstākļos tiek atklāts tikai piektdaļa bērnu līdz 12 mēnešu vecumam, kuri piedzimuši ar šī enzīma deficītu. Tāpēc vecākiem ir ļoti svarīgi pievērst uzmanību savu zīdaiņu aprūpei.
Turklāt agrīnas pazīmes ir acu sklēras un ausu un deguna skrimšļu pigmentācija (zilgani pelēka krāsa), ko bieži sauc par ohronozi.[ 10 ]
Laika gaitā parādās citi simptomi:
- izteikta ādas pigmentācija uz vaigu kauliem, padusēm un dzimumorgāniem;
- apģērba traipi, nonākot saskarē ar sviedrējošām ķermeņa zonām;
- vispārēja vājuma lēkmes;
- aizsmakusi balss.
Jāpatur prātā, ka alkaptonūrija un ohronoze, kā minēts iepriekš, ir sinonīmi nosaukumi vienam un tam pašam tirozīna katabolisma traucējumam.
Kļavu sīrupa urīna slimība un alkaptonūrija. Iedzimta kļavu sīrupa urīna slimība jeb leikinoze arī ir vielmaiņas traucējums, tai ir tāds pats iedzimtības modelis, un pat mutācijas rodas vienā hromosomā, bet ietekmē gēnu, kas kodē sazarotās ķēdes α-keto skābes dehidrogenāzes enzīmu kompleksu. Tā dēļ organisms nevar noārdīt noteiktus olbaltumvielu komponentus, jo īpaši aminoskābes leicīnu, izoleicīnu un valīnu. Ar šo slimību urīnam (un ausu sēram) ir salda smarža; turklāt šāda veida organiskās acidēmijas klīniskajā ainā ietilpst hipopigmentācija, asinsspiediena svārstības, krampji, vemšana un caureja, glikozes līmeņa pazemināšanās asinīs, ketoacidoze, halucinācijas utt. Mirstība bērniem ir diezgan augsta; pieaugušajiem bez ārstēšanas var iestāties koma un nāve smadzeņu tūskas dēļ.
Albinismu un alkaptonūriju “vieno” tikai tirozīns. Albinismu, ieskaitot okulokutānu, izraisa ģenētiskas mutācijas, kas ietekmē pigmenta melanīna ražošanu. Iedzimtas izmaiņas ir novērotas TYR gēnā 11. hromosomā (11q14.3), kas kodē tirozināzi, varu saturošu melanosomu enzīmu, kas nepieciešams ādas pigmenta veidošanai, pamatojoties uz tirozīna metabolisma produktiem. Šī slimība ir daudz biežāk sastopama nekā alkaptonūrija.
Komplikācijas un sekas
Alkaptonūrijas sekas un komplikācijas, ko izraisa tirozīna starpproduktu metabolīti – homogentisīnskābe un benzohinonetiķskābes –, rodas reaktīvu pigmentētu polimēru nogulsnēšanās, kolagēna fibrilu iznīcināšana un skrimšļa stāvokļa pasliktināšanās (samazinoties to izturībai pret mehānisko spriegumu).
Gadu gaitā, pieaugušā vecumā, attīstās lielo locītavu (gūžas, krustu un zarnkaula un ceļa locītavas) deģeneratīvs artrīts un osteoartrīts; sašaurinās starpskriemeļu telpas (īpaši jostas un krūšu kurvja mugurkaulā) – ar kalcifikāciju un osteofītu veidošanos; samazinās subhondrālo kaulu plātnīšu audu blīvums, un pamatā esošie kauli var patoloģiski pārveidoties, veidojoties izaugumiem un deformācijai. [ 11 ]
Var novērot sirds vārstuļu (aortas un mitrālā) un koronāro artēriju bojājumus – ar koronārās sirds slimības pazīmēm, kā arī akmeņu veidošanos nierēs un prostatas dziedzerī – tās pašas kalcifikācijas dēļ. [ 12 ], [ 13 ]
Diagnostika alkaptonūrija
Parasti iedzimtu vielmaiņas traucējumu diagnoze balstās uz organisma bioloģisko šķidrumu izpēti.
Pamatojoties uz kādiem testiem un reakcijām, var diagnosticēt alkaptonūriju? Lai noteiktu homogentisīnskābi un tās līmeni (norma – 20–30 mg dienā, paaugstināts – 3–8 g), ir nepieciešami urīna testi. Urīna paraugu pārbauda ar gāzu hromatogrāfiju vai masas spektrometriju, izmantojot šķidruma hromatogrāfiju; ir iespējams veikt skrīninga testu dzelzs hlorīda klātbūtnei urīnā. [ 14 ]
Ir arī ātras diagnostikas metode – alkaptona noteikšana izžuvušos urīna traipos uz papīra (pēc krāsas intensitātes).
Precizējot diagnozi, instrumentālā diagnostika (rentgenogrāfija) ietver osteoartrīta un citu locītavu patoloģiju radioloģisko pazīmju identificēšanu pacientiem.
Diagnozi apstiprina ar molekulārģenētiskām metodēm iedzimtu slimību diagnosticēšanai, piemēram, ģenētisko testēšanu un DNS sekvencēšanu. [ 15 ]
Diferenciālā diagnoze
Diferenciālā diagnoze ietver jaundzimušā hemohromatozi un akūtu aknu mazspēju, melaninūriju, akūtu intermitējošu porfīriju, hemofagocītu limfohistiocitozi, primāro mitohondriju patoloģiju, reimatoīdo artrītu, ankilozējošo spondilītu.
Kurš sazināties?
Profilakse
Gēnu mutāciju novēršana nav iespējama, taču, lai novērstu bērnu ar augstu iedzimtu defektu risku piedzimšanu, pastāv medicīniski ģenētiskā konsultēšana, kas ir nepieciešama pirms plānotas grūtniecības tiem pāriem, kuru ģimenes anamnēzē ir iedzimtas slimības. [ 19 ]
Prognoze
Alkaptonūrijas izraisīti letāli iznākumi ir ļoti reti, un nāvi var izraisīt nopietnas komplikācijas, kas saistītas ar sirdi un nierēm. Tāpēc kopējais paredzamais dzīves ilgums cilvēkiem ar alkaptonūriju ir labs.
Bet dzīves kvalitāte samazinās intensīvu locītavu vai mugurkaula sāpju dēļ ar ievērojamu mobilitātes ierobežojumu, bieži vien progresējošu.