Raksta medicīnas eksperts

Jaunas publikācijas
Xeroderma pigmentosum
Pēdējā pārskatīšana: 04.07.2025

Visi iLive saturs ir medicīniski pārskatīts vai pārbaudīts, lai nodrošinātu pēc iespējas lielāku faktisko precizitāti.
Mums ir stingras iegādes vadlīnijas un tikai saikne ar cienījamiem mediju portāliem, akadēmiskām pētniecības iestādēm un, ja vien iespējams, medicīniski salīdzinošiem pārskatiem. Ņemiet vērā, ka iekavās ([1], [2] uc) esošie numuri ir klikšķi uz šīm studijām.
Ja uzskatāt, ka kāds no mūsu saturiem ir neprecīzs, novecojis vai citādi apšaubāms, lūdzu, atlasiet to un nospiediet Ctrl + Enter.
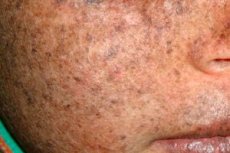
Xeroderma pigmentosum ir autosomāli recesīva ģenētiska DNS atjaunošanas slimība. Slimību izraisa mutācijas gēnos, kas ir iesaistīti ultravioletā starojuma un citu toksisku vielu bojātas DNS atjaunošanā.
Visbiežāk šī slimība skar bērnus, kurus sauc arī par "nakts bērniem". Biežas slimības komplikācijas ir bazālo šūnu karcinoma un citi ļaundabīgi ādas audzēji, metastātiska ļaundabīga melanoma un plakanšūnu karcinoma.
 [ 1 ]
[ 1 ]
Pathogenesis
Slimības attīstībā svarīgu lomu spēlē UV endonukleāzes enzīmu deficīts pacienta šūnās (fibroblastos) vai to pilnīga neesamība. Šis enzīms ir atbildīgs par ultravioleto staru bojātās DNS reprodukciju. Saskaņā ar citiem avotiem, dažiem pacientiem svarīgu lomu slimības attīstībā spēlē DNS polimerāzes-1 enzīma deficīts. Visbiežāk slimība attīstās staru ietekmē ar viļņa garumu 280-310 nm. Tiek uzskatīts, ka pigmenta kseroderma attīstās dažādu fotodinamisko vielu iekļūšanas organismā vai porfirīnu daudzuma palielināšanās dēļ cilvēka bioloģiskajā vidē.
Slimības sākumposmā novēro hiperkeratozi, epidermas fokusa atrofiju, Malpīgi slāņa retināšanu, melanīna granulu palielināšanos šūnu bazālajā slānī un hroniska iekaisuma infiltrāciju dermas augšējā slānī (galvenokārt ap asinsvadiem). Pēc tam tiek konstatētas deģeneratīvas izmaiņas kolagēna un elastīgajās šķiedrās, un audzēja stadijā atklājas ādas vēzim raksturīgas pazīmes.
Simptomi kseroderma pigmentosum
Šī slimība vienlīdz skar gan vīriešus, gan sievietes. Slimība sākas agrā bērnībā pavasarī vai vasarā. Pacienta āda ir īpaši jutīga pret saules gaismu. Tāpēc pirmie izsitumi hiperpigmentācijas veidā, līdzīgi kā dzimumzīmei, parādās uz ādas vietām, kas pakļautas saules gaismai. Izsitumi pastiprinās, eritēma pastiprinās, kļūst tumši brūna, parādās telangiektāzija, angioma un atrofija. Pēc tam aug papilomas un kārpas (galvenokārt uz sejas un kakla ādas), parādās plankumi un čūlas. Čūlu rētošanās rezultātā deguns kļūst plānāks ("putna knābis"), plakstiņš izvirzās uz āru. Papilomas parasti attīstās ļaundabīgos audzējos. Pigmentēta kseroderma, kā likums, rodas kopā ar spinocelulāru vēzi, melanosarkomām.
Kas tevi traucē?
Posmi
Kseroderma pigmentosum klīniskajā gaitā izšķir piecus posmus: iekaisuma (eritematozo), hiperpigmentāciju, atrofiju, hiperkeratozi un ļaundabīgos audzējus.
Iekaisuma (eritematozās) stadijas laikā uz ādas vietām, kas pakļautas saules gaismai (sejas, kakla, krūšu kurvja augšdaļas, roku, plaukstu), parādās pietūkums, sarkani plankumi un dažreiz pūslīši un pūslīši. Vietās, kas nav pakļautas saules gaismai, izsitumu gandrīz nav.
Ar hiperpigmentāciju sarkano plankumu vietā parādās gaiši brūni, brūni hiperpigmentēti plankumi, kas līdzīgi dzimumzīmēm.
Atrofijas gadījumā āda izžūst, kļūst plānāka un grumbaina. Uz lūpu un deguna ādas novēro daudzas mazas, zvaigžņveida telangiektāzijas un rētas ar spīdīgu virsmu. Atrofijas un rētu dēļ attīstās mikrotomija (mutes atveres samazināšanās), ektropions, ausu un deguna retināšana, deguna atveres un mutes atrēzija. 80–85% pacientu tiek skartas acis: tiek atzīmēts konjunktivīts, keratīts, radzenes un gļotādas bojājumi un redzes pavājināšanās. Uz plakstiņu ādas novēro dishromiju, telangiektāziju, hiperkeratotiskas izmaiņas un audzējus.
Ar hiperkeratozi iepriekš aprakstītajos patoloģiskajos perēkļos parādās kārpaini audzēji, papiloma, keratoakantoma, fibroma, angiofibrioma un citi labdabīgi audzēji. Tāpēc daži zinātnieki pigmenta kserodermu iekļauj pirmsvēža slimību grupā.
Pēc 10–15 gadiem no slimības sākuma atrofiski izmainītos perēkļos un pigmenta plankumos parādās ļaundabīgi ādas audzēji (bazalioma, endotelioma, angiosarkoma). Audzēji īsā laikā iznīcina, metastazējas iekšējos orgānos un noved pie nāves. Dažu pacientu iekšējos orgānos un audos novērojamas vispārējas distrofiskas izmaiņas (otrā un trešā pirksta sindaktēlijas, zobu distrofija, pilnīgs matu izkrišana utt.).
Veidlapas
Xeroderma pigmentosum neiroloģiskā forma izpaužas divos sindromos.
Rīda sindromu raksturo kserodermas pigmentosuma un mikrocefālijas klīniskās izpausmes, idiopātija un lēna skeleta sistēmas augšana. Slimības neiroloģiskajā formā DNS reparācija ir sarežģīta, un rentgena terapija izraisa galveno patoloģisko perēkļu palielināšanos.
De Sinctis-X Cocchione sindromu raksturo šādas klīniskās pazīmes:
- kserodermas pigmentosum klīnisko pazīmju attīstība uz īpaši gaismjutīgas ādas;
- ļaundabīgo audzēju agrīna parādīšanās;
- vienlaicīga spastiskās paralīzes, mikrocefālijas un demences gaita;
- iedzimtas deformācijas un pundurisms;
- dzimumdziedzeru nepietiekama attīstība;
- biežas spontānās aborts;
- recesīva pārnešana mantojuma ceļā.
Pēc dažu dermatologu domām, Santis-Cacchione sindroms nav patstāvīga slimība, bet gan smaga un pilnībā izteikta kseroderma pigmentosum klīniskā izpausme.
Ģenētiskās formas
Veidi |
Gēns |
Lokuss |
Apraksts |
A, I tips, XPA |
XPA |
9q22.3 |
Xeroderma pigmentosum (XP) A grupa – klasiskā forma |
B tips, II, XPB |
XPB |
2q21 |
XP B grupa |
C tips, III, XPC |
XPC |
3p25 |
XP C grupa |
D, IV, XPD tips |
XPD ERCC6 |
19q13.2–q13.3, 10q11 |
XP D grupa vai De Sanctis-Cacchione sindroms |
E, V, XPE tips |
DDB2 |
11. lpp. 12–11 |
XP E grupa |
F, VI, XPF tips |
ERCC4 |
16. 13.3.–13.13. lpp. |
XP F grupa |
G, VII, XPG tips |
RAD2ERCC5 |
13q33 |
XP G grupa un COFS sindroms (cerebro-okulo-facioskeletālais sindroms) 3. tips |
V tips, XPV |
POLH |
6. lpp. 21.1–12. |
Variants Xeroderma Pigmentosa |
Kas ir jāpārbauda?
Kā pārbaudīt?
Kurš sazināties?
Prognoze
Lielākā daļa pacientu (2/3) mirst pirms 15 gadu vecuma sasniegšanas. Daži pacienti var nodzīvot līdz 40–50 gadiem.
[ 20 ]