Raksta medicīnas eksperts

Jaunas publikācijas
Polijas sindroms
Last reviewed: 04.07.2025

Visi iLive saturs ir medicīniski pārskatīts vai pārbaudīts, lai nodrošinātu pēc iespējas lielāku faktisko precizitāti.
Mums ir stingras iegādes vadlīnijas un tikai saikne ar cienījamiem mediju portāliem, akadēmiskām pētniecības iestādēm un, ja vien iespējams, medicīniski salīdzinošiem pārskatiem. Ņemiet vērā, ka iekavās ([1], [2] uc) esošie numuri ir klikšķi uz šīm studijām.
Ja uzskatāt, ka kāds no mūsu saturiem ir neprecīzs, novecojis vai citādi apšaubāms, lūdzu, atlasiet to un nospiediet Ctrl + Enter.
Reta ķermeņa struktūras intrauterīnās veidošanās anomālija, kas galvenokārt sastāv no krūšu muskuļa krūšu kaula un ribu daļas hipoplāzijas vai tās pilnīgas neesamības. Tā nosaukta angļu ķirurga vārdā, kurš aprakstīja paraugu ar ribu muskuļu defektu, ar kuru viņš saskārās, strādājot nepilnu slodzi morgā vēl studenta gados. A. Polands nebija pirmais; pirms viņa atsevišķi gadījumi jau 19. gadsimta sākumā bija piesaistījuši uzmanību Francijā un Vācijā, taču tieši viņa publikācija iezīmēja nopietnas šīs iedzimtās patoloģijas izpētes sākumu. 20. gadsimta sākumā Dž. Tomsons publicēja pilnīgu šīs slimības aprakstu. Kopš tā laika pasaules medicīnas literatūrā ir aprakstīti aptuveni 500 šādu gadījumu.
 [
[ Epidemioloģija
Saslimstības statistika liecina, ka iedzimtas ribu muskuļu anomālijas, kas izteiktas dažādās pakāpēs, rodas vidēji vienam jaundzimušajam no 30 tūkstošiem vai nedaudz vairāk dzīvi dzimušiem bērniem. Zēni ar šādiem attīstības defektiem piedzimst biežāk.
Līdz pat 80% deformāciju Polijas sindromā ir labās puses. Traucējumi ir izteikti dažādās pakāpēs, un nav atbilstības starp krūškurvja un rokas veidošanās anomāliju smagumu.
Cēloņi Polijas sindroms
Bērnu ar šo anomāliju dzimšanas cēloņi līdz šim joprojām ir hipotētiski. Mantojuma veids un gēns, kas pārnes šo patoloģiju, nav noteikti, taču ir aprakstīti reti ģimenes anamnēzes gadījumi ar Pola sindromu. Tiek pieņemta recesīva mantošana. Tiek uzskatīts, ka slimības pārnešanas varbūtība no slima vecāka uz saviem bērniem ir aptuveni 50%. Lielākā daļa gadījumu ir vientuļi. Bērnu ar šo anomāliju dzimšanas riska faktori ir ārēja un iekšēja teratogēna ietekme uz embriju dēšanas un orgānu un sistēmu attīstības periodā. Pastāv vairākas hipotēzes, kas izskaidro šī ribu-muskuļu defekta etioloģiju un patogenēzi, taču neviena no tām nav pilnībā apstiprināta. Visticamākais pieņēmums ir, ka kāds nelabvēlīgs faktors provocē embrija asinsapgādes nepietiekamību sestajā grūtniecības nedēļā, kad veidojas zematslēgas artērija. Tas izraisa tās nepietiekamu attīstību (lūmena sašaurināšanos) un nepietiekamu asinsapgādi, kas noved pie lokālas mīksto audu un kaulu hipoplāzijas. Bojājuma apmēru nosaka artērijas un/vai tās zaru bojājuma pakāpe.
Iemesli ietver arī embrija krūšu kurvja ribu muskuļu audu šūnu migrācijas traucējumus vai to intrauterīnu traumu. Tomēr nevienai no šīm hipotēzēm līdz šim nav pietiekamu ticamu pierādījumu.
Simptomi Polijas sindroms
Pirmās šīs iedzimtās anomālijas pazīmes ir vizuāli pamanāmas jau zīdaiņa vecumā pēc krūšu muskuļa un paduses raksturīgā izskata. Un rokas hipoplāzijas klātbūtnē - no dzimšanas brīža.
Sindroma simptomu komplekss ir šāds:
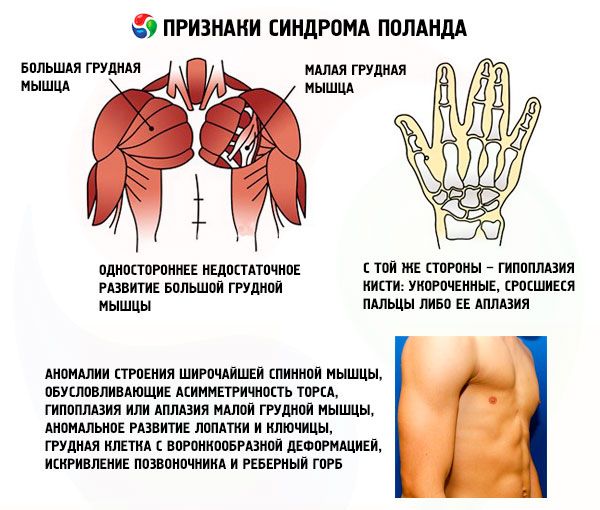
- vienpusēja nepietiekama krūšu kurvja muskuļa vai tā fragmentu attīstība, visbiežāk krūšu kurvja un ribu muskuļi;
- vienā pusē – rokas hipoplāzija: saīsināti, sapluduši pirksti vai to aplazija; nepietiekama piena dziedzeru attīstība vai to neesamība, atēlija; zemādas tauku slāņa retināšanās; padušu apmatojuma neesamība; skrimšļa/kaula ribu audu struktūras anomālijas vai to pilnīga neesamība (parasti III un IV).
Visu otrajā daļā aprakstīto zīmju klātbūtne nav obligāta; tās var kombinēt ar pirmo dažādos veidos.
Papildus uzskaitītajām var rasties ārkārtīgi retas latissimus dorsi muskuļa struktūras anomālijas, kas izraisa rumpja asimetriju, minora krūšu muskuļa hipoplāziju vai aplaziju, lāpstiņas un atslēgas kaula patoloģisku attīstību, piltuvveida krūtis, mugurkaula izliekumu un ribu kupri.
Ar kreiso defektu bieži tiek novērota iekšējo orgānu transpozīcija, jo īpaši sirds ir nobīdīta pa labi. Ar normālu sirds atrašanās vietu kombinācijā ar ribu neesamību tā ir praktiski neaizsargāta, un tās pukstēšana ir pamanāma zem ādas.
Polijas sindroms bērniem parasti ir pamanāms jau no dzimšanas brīža, bet dažos gadījumos nelieli defekti netiek atklāti līdz aptuveni trīs gadu vecumam.
Saskaņā ar lokalizāciju krūškurvja strukturālo elementu defekti tiek sadalīti priekšējās, aizmugurējās un sānu sienu deformācijās.
Polijas sindroms meitenēm pubertātes laikā, pat visvieglākajā formā, izpaužas ar to, ka krūts bojātajā pusē neveidojas vai atpaliek attīstībā un atrodas ievērojami augstāk nekā normālajā pusē. Vieglos slimības gadījumos zēniem sindroms dažreiz tiek atklāts diezgan vēlu, pusaudža gados, kad nav iespējams "uzpumpēt" muskuli bojātajā pusē.
Polijas sindroms sievietēm neietekmē hormonālo līmeni vai spēju ieņemt bērnu.
Vairumā gadījumu Polijas sindroms ir kosmētisks defekts: visbiežāk krūšu muskulis ir deformēts vai tā nav, nav krūškurvja defekta un ir pilnībā funkcionējoša roka. Augšējo ekstremitāšu motoriskās funkcijas ir saglabātas, un nekas neliedz šādiem pacientiem intensīvi nodarboties ar sportu.
Tomēr pastāv arī citi, traumatiskāki šīs patoloģijas veidi. Šādu gadījumu sekas un komplikācijas ir nedaudz nopietnākas. Atkarībā no deformāciju smaguma pakāpes pacientam var attīstīties elpošanas un hemodinamikas traucējumi. Kostohondrālā karkasa pilnīgas neesamības gadījumos parasti tiek konstatēta plaušu trūce, un elpošanas traucējumi izpaužas jau no dzimšanas brīža.
Retāko kreisās puses patoloģiju gadījumos kombinācijā ar ribu trūkumu ar normālu orgānu izvietojumu sirds atrodas tieši zem ādas.Šāda pacienta dzīvību pastāvīgi apdraud briesmas, kas saistītas ar iespējamu traumu un sirdsdarbības apstāšanos.
Bērnam ar izteiktu krūškurvja defektu parasti ir problēmas ar hemodinamiku, jo samazinās sistoliskais un palielinās diastoliskais arteriālais spiediens kombinācijā ar paaugstinātu venozo spiedienu. Šādiem bērniem raksturīgs paaugstināts nogurums, astēnisks sindroms, viņi var atpalikt no saviem vienaudžiem fiziskajā attīstībā.
Polijas sindroma izpausmes attiecas arī uz subklāvijas artērijas un/vai tās zaru struktūru, kas rada apstākļus arteriālās asinsrites traucējumiem defekta pusē.
Tiek novērotas dažas svarīgu iekšējo orgānu struktūras un izvietojuma anatomiskas anomālijas. To izpausmes pakāpe var ievērojami sarežģīt pacienta stāvokli. Tā ir sirds novirze no normālā stāvokļa vienā vai otrā virzienā līdz transpozīcijai, tās robežu paplašināšanai vai rotācijai pulksteņrādītāja virzienā, plaušu un nieru hipoplāzija defektīvajā pusē.
Posmi
Šajā slimībā var izdalīt četrus krūšu veidošanās posmus.
Pirmais ir raksturīgs lielākajai daļai zināmo gadījumu, kad tikai mīkstie audi ir patoloģiski attīstīti, un krūškurvja forma un ribu skrimšļaino un kaulaino daļu struktūra ir normāla.
Otrais ir tad, kad deformācijas skar krūtis: defektīvā puse, kurā saglabājušās ribu kaula un skrimšļa daļas, ir nedaudz iespiesta ribu skrimšļu rajonā, krūšu kauls ir pagriezts uz pusi uz sāniem, bet pretējā pusē bieži novēro izvirzītu (ķīļveida) krūšu daļu.
Trešajā posmā ribu kaula daļas struktūra ir saglabājusies, bet skrimšļa daļa ir nepietiekami attīstīta, krūškurvis ir asimetrisks, krūšu kauls ir slīps deformācijas virzienā, bet nav konstatētas rupjas anomālijas.
Ceturto posmu raksturo ribu skrimšļainās un kaulainās daļas neesamība no vienas līdz četrām (no III līdz VI). Bojātajā pusē trūkstošo ribu vietā ir ieplaka, krūšu kauls ir manāmi pagriezts.
Tomēr jebkurā krūškurvja struktūras elementu veidošanās posmā bērna ķermeņa stāvoklis var būt normāls (kompensēts), ar periodiskiem uzlabojumiem (subkompensēts) un ar pieaugošu iekšējo orgānu un skeleta sistēmas darbības pasliktināšanos (dekompensēts). Tas ir atkarīgs no organisma individuālajām īpašībām, attīstības ātruma, komorbiditātes un dzīvesveida.
Diagnostika Polijas sindroms
Iedzimta ribu-muskuļu patoloģija tiek noteikta vizuāli, ārsts palpē pacientu un izraksta rentgenu. Tas parasti ir pietiekami, lai noteiktu krūškurvja bojājuma pakāpi un veidu. Precīzāku priekšstatu par slimību var sniegt datortomogrāfija un magnētiskās rezonanses attēlveidošana.
Tiek nozīmēta arī subklāvijas artērijas ultraskaņas izmeklēšana, lai noteiktu tās diametru, smadzeņu ultraskaņas izmeklēšana un cita instrumentālā diagnostika, kā norādīts.
Lai novērtētu vienlaicīgus anatomiskus defektus, nepieciešama kardiologa konsultācija un elektrokardiogrāfija, sirds ultraskaņas izmeklēšana, veloergometrija, ehokardiogrāfija un galveno asinsvadu doplerogrāfija.
Elpošanas traucējumu gadījumā nepieciešama pulmonologa konsultācija, kas var nozīmēt plaušu funkcionālā stāvokļa izpēti, piemēram, spirogrāfiju.
Šīs slimības testu rezultāti parasti ir normas robežās, ja vien nav vienlaicīgu patoloģiju.
Rūpīgi diagnostikas pasākumi ļauj precīzi novērtēt rekonstruktīvās iejaukšanās apjomu.
Kādi testi ir vajadzīgi?
Prognoze
Vairumā gadījumu šis iedzimtais ķermeņa struktūras defekts ir reducēts līdz muskuļu nepietiekamai attīstībai, kas neietekmē ekstremitāšu un iekšējo orgānu darbību, sieviešu auglību un ir noņemams kosmētiskais defekts. Pat ar sarežģītākiem kombinētiem bojājumiem savlaicīga ārstēšana garantē pacientam iespēju dzīvot pilnvērtīgu dzīvi.