Raksta medicīnas eksperts

Jaunas publikācijas
Ushera sindroms
Pēdējā pārskatīšana: 04.07.2025

Visi iLive saturs ir medicīniski pārskatīts vai pārbaudīts, lai nodrošinātu pēc iespējas lielāku faktisko precizitāti.
Mums ir stingras iegādes vadlīnijas un tikai saikne ar cienījamiem mediju portāliem, akadēmiskām pētniecības iestādēm un, ja vien iespējams, medicīniski salīdzinošiem pārskatiem. Ņemiet vērā, ka iekavās ([1], [2] uc) esošie numuri ir klikšķi uz šīm studijām.
Ja uzskatāt, ka kāds no mūsu saturiem ir neprecīzs, novecojis vai citādi apšaubāms, lūdzu, atlasiet to un nospiediet Ctrl + Enter.
Ašera sindroms ir iedzimta slimība, kas izpaužas kā pilnīgs kurlums no dzimšanas brīža, kā arī progresējoša aklums ar vecumu. Redzes zudums ir saistīts ar pigmentozu retinītu — tīklenes pigmentāras deģenerācijas procesu. Daudziem cilvēkiem ar Ašera sindromu ir arī smagas līdzsvara problēmas.
Epidemioloģija
Pateicoties pētījumam, bija iespējams konstatēt, ka Ašera sindroms skar aptuveni 8% no pārbaudītajiem kurlmēmajiem bērniem (testi tika veikti īpašās iestādēs kurlmēmajiem cilvēkiem). Pigmentārs retinīts tika novērots 6–10% pacientu, kas cieš no iedzimta kurluma, kas, savukārt, novērojams aptuveni 30% cilvēku ar pigmentāras tīklenes slimību.
Tiek uzskatīts, ka šī slimība izpaužas aptuveni 3–10 cilvēkiem no 100 tūkstošiem visā pasaulē. To var vienlīdz novērot gan sievietēm, gan vīriešiem. No šī sindroma cieš aptuveni 5–6% pasaules iedzīvotāju. Aptuveni 10% no visiem bērnības dziļas kurlības gadījumiem rodas Ašera sindroma I, kā arī II tipa dēļ.
Amerikas Savienotajās Valstīs visbiežāk sastopamie ir 1. un 2. tips. Kopā tie veido aptuveni 90 līdz 95 procentus no visiem Ašera sindroma gadījumiem bērniem.
Cēloņi Ushera sindroms
Ašera sindroma I, II un III tipam ir autosomāli recesīvs cēlonis, savukārt IV tips tiek uzskatīts par X hromosomu slimību. Akluma un kurluma cēloņi, kas rodas šī sindroma gadījumā, vēl nav pietiekami pētīti. Tiek pieņemts, ka cilvēkiem ar šo slimību ir paaugstināta jutība pret komponentiem, kas var bojāt DNS struktūru. Turklāt šī slimība var būt saistīta ar imūnsistēmas traucējumiem, taču šajā gadījumā nav precīza priekšstata par šo procesu.
1989. gadā pacientiem ar II tipa slimību pirmo reizi tika identificētas hromosomu anomālijas, kas nākotnē varētu novest pie sindroma izraisītāju gēnu izolēšanas. Iespējams, ka šos gēnus būs iespējams identificēt arī nesējiem un izstrādāt īpašus pirmsdzemdību ģenētiskos testus.
 [ 8 ]
[ 8 ]
Riska faktori
Sindroms tiek mantots, ja ir skarti abi vecāki, t.i., tas tiek mantots recesīvā veidā. Bērns var mantot šo slimību arī tad, ja viņa vecāki ir gēna nesēji. Ja abiem nākamajiem vecākiem ir šis gēns, tad varbūtība, ka bērnam piedzims šis sindroms, ir 1 no 4. Persona, kurai ir tikai viens sindroma gēns, tiek uzskatīta par nesēju, bet viņai nav šīs slimības simptomu. Mūsdienās vēl nav iespējams noteikt, vai cilvēkam ir šīs slimības gēns.
Ja bērns piedzimst vecākiem, no kuriem vienam nav šāda gēna, tad varbūtība, ka viņš mantos sindromu, ir ļoti zema, taču viņš noteikti būs nesējs.
Simptomi Ushera sindroms
Ašera sindroma simptomi ir dzirdes zudums un pigmentētu šūnu patoloģiska uzkrāšanās acu struktūrās. Pēc tam pacientam attīstās tīklenes deģenerācija, kas izraisa redzes pasliktināšanos un smagākajos gadījumos - redzes zudumu.
Sensorineirāls dzirdes zudums var būt viegls vai pilnīgs, un parasti tas neprogresē no dzimšanas brīža. Tomēr tīklenes pigmenta slimība var sākt attīstīties bērnībā vai vēlāk. Testu rezultāti liecina, ka centrālo redzes asumu var saglabāt daudzus gadus, pat ja perifērā redze pasliktinās (stāvoklis, ko sauc par "tuneļa redzi").
Šīs ir galvenās slimības izpausmes, kuras dažkārt var papildināt ar citiem traucējumiem, piemēram, psihozi un citiem garīgiem traucējumiem, problēmām ar iekšējo ausi un/vai kataraktu.
Veidlapas
Pētījuma laikā tika identificēti 3 šīs slimības veidi, kā arī 4. forma, kas ir diezgan reta.
I tipa slimībai raksturīga iedzimta pilnīga kurlība, kā arī līdzsvara traucējumi. Bieži vien šādi bērni sāk staigāt tikai 1,5 gadu vecumā. Redzes pasliktināšanās parasti sākas 10 gadu vecumā, un nakts akluma stāvokļa galīgā attīstība sākas 20 gadu vecumā. Bērniem ar šāda veida slimību var attīstīties progresējoša perifērās redzes pasliktināšanās.
II tipa slimības gadījumā novēro mērenu vai iedzimtu kurlumu. Šajā gadījumā daļēja kurluma pasliktināšanās bieži vien vairs nenotiek. Pigmentārais retinīts sāk attīstīties aptuveni pusaudža vecuma beigās vai pēc 20 gadiem. Nakts akluma attīstība parasti sākas 29–31 gada vecumā. Redzes asuma traucējumi II tipa patoloģijas gadījumā parasti progresē nedaudz lēnāk nekā I tipa patoloģijas gadījumā.
Slimības III tipu raksturo progresējošs dzirdes zudums, kas parasti sākas pubertātes laikā, kā arī pakāpeniska pigmentozas retinīta attīstība tajā pašā periodā (nedaudz vēlāk nekā dzirdes zudums), kas var kļūt par faktoru progresējošas akluma attīstībā.
IV tipa patoloģijas izpausmes galvenokārt rodas vīriešiem. Šajā gadījumā tiek novēroti arī progresējoši traucējumi un dzirdes un redzes zudums. Šī forma ir ļoti reta un parasti tai ir X hromosomu raksturs.
Diagnostika Ushera sindroms
Ašera sindroma diagnoze tiek noteikta, pamatojoties uz pacienta novēroto pēkšņas kurlības un progresējoša redzes zuduma kombināciju.
Testi
Mutācijas noteikšanai var tikt nozīmēts īpašs ģenētiskais tests.
Ir atrasti vienpadsmit ģenētiskie lokusi, kas var izraisīt Ašera sindroma attīstību, un ir identificēti deviņi gēni, kas noteikti ir šī traucējuma cēlonis:
- 1. tips: MY07A, USH1C, Cdh23, Pcdh15, SANS.
- 2. tips: ush2a, VLGR1, WHRN.
- Ašera sindroma 3. tips: USH3A.
NIDCD zinātnieki kopā ar kolēģiem no universitātēm Ņujorkā un Izraēlā ir identificējuši mutāciju ar nosaukumu R245X Pcdh15 gēnā, kas veido lielu daļu 1. tipa Ašera sindroma ebreju populācijā.
Lai uzzinātu par laboratorijām, kas veic klīniskos pētījumus, apmeklējiet vietni https://www.genetests.org un laboratoriju direktorijā meklējiet "Usher sindroms".
Lai uzzinātu par esošajiem klīniskajiem pētījumiem, kas ietver Ašera sindroma ģenētisko testēšanu, apmeklējiet vietni https://www.clinicaltrials.gov un meklējiet "Ašera sindroms" vai "Ašera sindroma ģenētiskā testēšana".
[ 25 ], [ 26 ], [ 27 ], [ 28 ], [ 29 ], [ 30 ]
Instrumentālā diagnostika
Ir vairākas instrumentālās diagnostikas metodes:
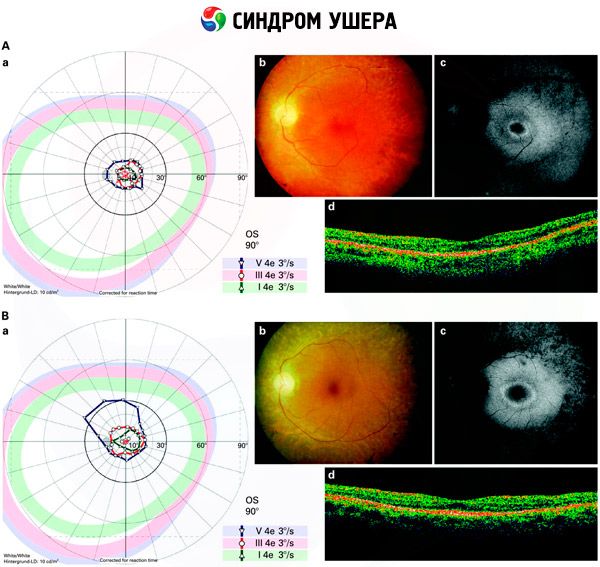
- Acs dibena pārbaude, lai noteiktu pigmenta plankumu klātbūtni tīklenē, kā arī tīklenes asinsvadu sašaurināšanos;
- Elektroretinogramma, kas ļauj noteikt sākotnējās deģeneratīvās novirzes acs tīklenē. Tā parāda elektroradiogrāfisko ceļu izzušanu;
- Elektronistagmogramma (ENG) mēra nekontrolētas acu kustības, kas varētu liecināt par nelīdzsvarotības klātbūtni.
- Audiometrija, ko izmanto, lai noteiktu kurluma klātbūtni un tā smagumu.
Diferenciālā diagnoze
Ašera sindroms ir jānošķir no dažiem līdzīgiem traucējumiem.
Hallgrena sindroms, kam raksturīgs iedzimts dzirdes zudums un progresējošs redzes zudums (attīstās arī katarakta un nistagms). Papildu simptomi ir ataksija, psihomotoriski traucējumi, psihoze un garīga atpalicība.
Alstroma sindroms, kas ir iedzimta slimība, kuras gadījumā deģenerējas tīklene, kā rezultātā tiek zaudēta centrālā redze. Šis sindroms ir saistīts ar bērnu aptaukošanos. Vienlaikus pēc 10 gadiem sāk attīstīties cukura diabēts un dzirdes zudums.
Masaliņas grūtniecei pirmajā trimestrī var izraisīt dažādas novirzes bērna attīstībā. Starp šādas anomālijas sekām ir dzirdes zudums, kā arī (vai) redzes problēmas, un papildus tam dažādi attīstības defekti.
Kurš sazināties?
Prognoze
Ašera sindromam ir nelabvēlīga prognoze. Vairumam pacientu ar šo jebkāda veida slimību redzes lauks un tā asums sāk pasliktināties 20–30 gadu laikā. Dažos gadījumos rodas pilnīgs divpusējs redzes zudums. Dzirdes zudums, ko vienmēr pavada mēmums, ļoti ātri attīstās līdz pilnīgam divpusējam dzirdes zudumam.